Y-mer:一种基于k-mer用于从超低测序深度数据确定人类Y染色体单倍群的方法
https://doi.org/10.21203/rs.3.rs-5042960/v2 # 摘要 对于保存不佳或混合样本,由于目标位点只能获得超低测序深度(ulcWGS)< 0.1x的序列,确定个体的遗传祖先具有挑战性。利用最近在使用长读长进行端到端全基因组测序方面的进展,我们开发了一种新的基于k-mer的方法Y-mer,并展示了如何在基于距离的模型中利用数十万个k-mers的信息,在测序深度低于0.01x的情况下,无需额外的PCR或捕获即可准确推断chrY单倍群。我们在古DNA和产前筛查数据上测试了Y-mer的性能,展示了其在从短读长WGS数据中进行无细胞DNA、法医和古DNA研究的遗传祖先
https://doi.org/10.21203/rs.3.rs-5042960/v2
摘要
对于保存不佳或混合样本,由于目标位点只能获得超低测序深度(ulcWGS)< 0.1x的序列,确定个体的遗传祖先具有挑战性。利用最近在使用长读长进行端到端全基因组测序方面的进展,我们开发了一种新的基于k-mer的方法Y-mer,并展示了如何在基于距离的模型中利用数十万个k-mers的信息,在测序深度低于0.01x的情况下,无需额外的PCR或捕获即可准确推断chrY单倍群。我们在古DNA和产前筛查数据上测试了Y-mer的性能,展示了其在从短读长WGS数据中进行无细胞DNA、法医和古DNA研究的遗传祖先推断方面的潜力。
关键词:古DNA、超低测序深度全基因组测序、无创产前筛查、无创产前检测、k-mer、人类、Y染色体、单倍群、预测
引言
由于Y染色体的高度复杂性,它是人类基因组中最后一个被完全测序的染色体(Rhie等,2023)。其长度约一半由常染色质、与X染色体重组区域(PAR1和PAR2)以及与人类X染色体不重组的区域组成,而另一半包括着整个人类基因组中最大的着丝粒和异染色质区域(Yq12),这些区域直到最近Y染色体组装完成前一直未被完全测绘(Rhie等,2023)。
Skov等人(Skov和Schierup,2017)对62个丹麦Y染色体进行高覆盖度测序研究,使用了多种文库插入大小,以及Esteller-Cucala等人(Esteller-Cucala等,2023)对7个主要Y染色体单倍群的Y染色体组装研究,揭示了参考基因组(GRCh38)序列中存在重大空白,并发现着丝粒区域存在高度变异性,以及某些单倍群特有的结构变异。
此外,最近两项使用高覆盖度长读长测序数据来确定端到端Y染色体序列的研究揭示,扩增子基因数量高度可变,Y染色体总长度差异可达两倍(Hallast等,2023; Rhie等,2023)。Y染色体长度变异的主要来源是Yq12区域,在43个已完全测序的Y染色体样本中,该区域不仅在不同单倍群之间,而且在两个最常见的单倍群E1和O2内部都表现出高度的长度变异(Hallast等,2023)。然而,某些重复区域(如TSPY阵列)表现出受系统发育限制的长度变异。Y染色体序列各个组成块的大小差异在多大程度上具有系统发育信息价值,还需要通过更大样本量的系统研究来确定。
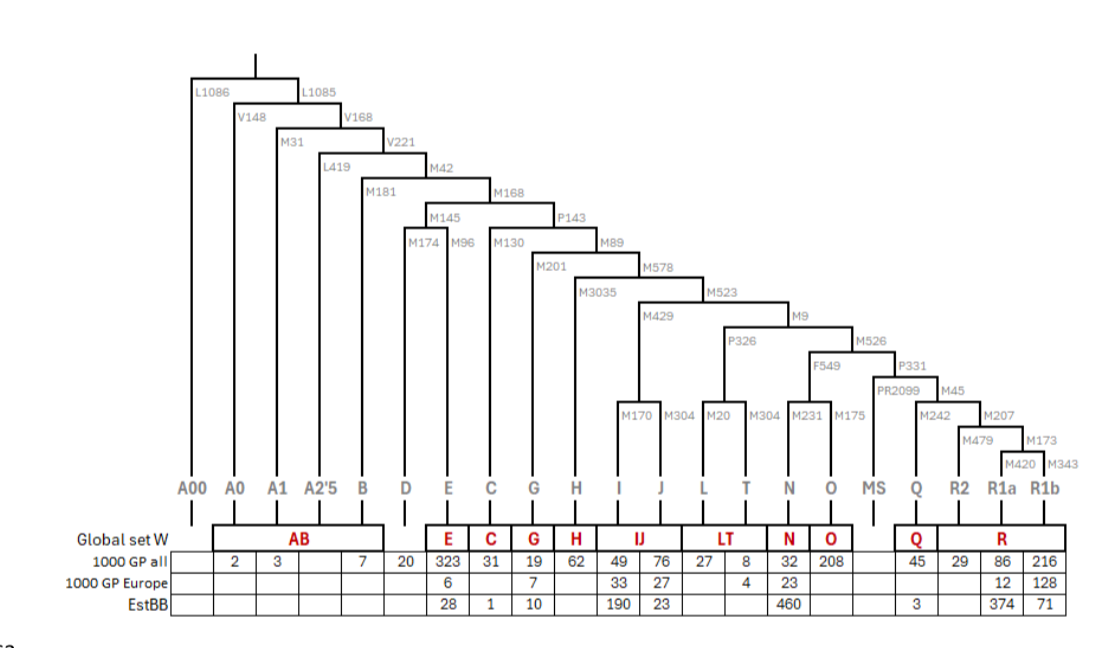
人类Y染色体单倍群被定义为基于非重组区域变异所绘制的系统发育树中的分支或支系。这些单倍群通常用字母数字标记,基本单倍群结构如图1所示,更详细的亚支系版本见表S4。Y染色体单倍群常规用于古DNA(aDNA)研究,因为它们能指示特定于男性的基因流动,并且对于阐明从常染色体数据中已确定存在亲缘关系的个体之间的具体关系很重要。它们在法医实践中对个体识别也具有相关性。然而,当测序深度低于0.1×特别是低于0.01×时,从散弹测序数据中确定Y染色体单倍群具有挑战性,因为Y系统发育中的大多数分支仅由不到10个SNV定义,只有少数分支由>100个SNV定义。对于保存不佳的样本,由于DNA高度片段化、损坏或内源DNA比例低,通过捕获或额外的散弹测序来增加测序深度可能不切实际。
图1. Y染色体单倍群的示意系统发育树。树下方显示了来自1000基因组计划(1000 GP)的1243个个体、其欧洲亚集的240个个体,以及爱沙尼亚生物银行(EstBB)的1160个个体中各单倍群的个体数量。
在本研究中,我们探索了从超低测序深度全基因组测序(ulcWGS)数据预测Y染色体单倍群的潜力。
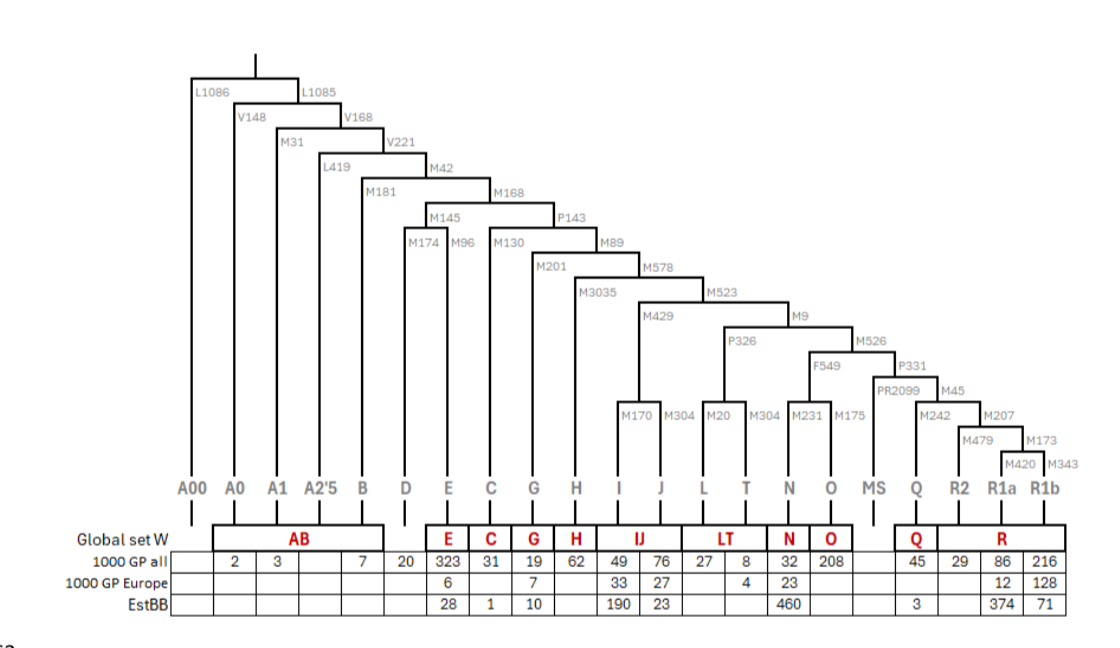
图1. Y染色体单倍群的示意系统发育树。树下方显示了1000基因组计划(1000 GP)的1243个个体、其欧洲亚集的240个个体,以及爱沙尼亚生物银行(EstBB)的1160个个体中各单倍群的个体数量。
在本研究中,我们使用k-mers探索从ulcWGS数据预测Y染色体单倍群的潜力。为此,我们开发了一个新工具Y-mer,该工具使用人类Y染色体完整序列特有的k-mers计数。我们首先通过对1000基因组计划(Auton等,2015)和爱沙尼亚生物银行(EstBB)高覆盖度序列(Milani等,2025)中已知单倍群的Y染色体进行降采样,来查询所需的最佳k-mers数量并确定该方法的序列覆盖度下限。我们使用区域和全球样本的训练集开发了Y-mer的低分辨率和高分辨率单倍群预测模型,以实现该工具的广泛用途,并在相关验证集中确定预测准确性。最后,我们在三个区域研究的超低测序深度古DNA和来自爱沙尼亚(Žilina等,2019)和中国(Xu等,2018)的无创产前筛查(NIPS)数据上测试了该方法。
结果
基于距离的模型预测多人群数据中的Y染色体单倍群
由于我们的目标是开发一种基于k-mer从ulcWGS数据确定Y染色体单倍群的方法,我们需要知道用于可靠预测基本单倍群所需的最少k-mer数量和源Y染色体组装。此外,由于我们构建了基于距离的单倍群预测模型(图2),其中比较了竞争单倍群之间的k-mer频率,我们想知道在基本单倍群上准确工作的方法是否可以扩展到更多的亚支系,以及在人群A的训练集上开发的单倍群模型是否可以成功应用于人群B的个体。我们还想知道这种方法对噪声的鲁棒性如何,以及在古DNA的情况下对污染的抵抗力如何。
首先,为了探索从全球多个人群中可靠预测chrY单倍群所需的chrY k-mers数量及其源基因组,我们生成并测试了基于数十万到数百万k-mers的模型。我们使用五组不同的高覆盖度基因组作为chrY数据的来源创建了模型(图2),第一组仅使用单个chrY(HG002,表S1)T2T长读长组装,第二组使用来自不同单倍群的21个T2T Y染色体组装(Hallast等,2023,表S2),最后三组使用来自1000 GP(Auton等,2015)和EstBB数据(Milani等,2025)的110-222个Y染色体的短读长数据。使用GenomeTester4的glistmaker(Kaplinski,Lepamets和Remm,2015),我们提取了特定于chrY的k-mers列表,包括多达1400万个25-mers(详见材料和方法,表S3)。接下来,我们从这些列表中提取了10,000到100,000个k-mers集合,以确定所需的最小k-mers数量。我们在三个不同的地理范围内进行了单倍群预测训练,W集代表11个全球单倍群,E集代表欧洲常见的22个单倍群,NE集代表东北欧特有的23个单倍群(图2,表S4,S5)。在每个模型中,单倍群预测是通过基于距离的聚类完成的,考虑模型中所有k-mers,将个体分配给距离最小的单倍群(附录模型)。
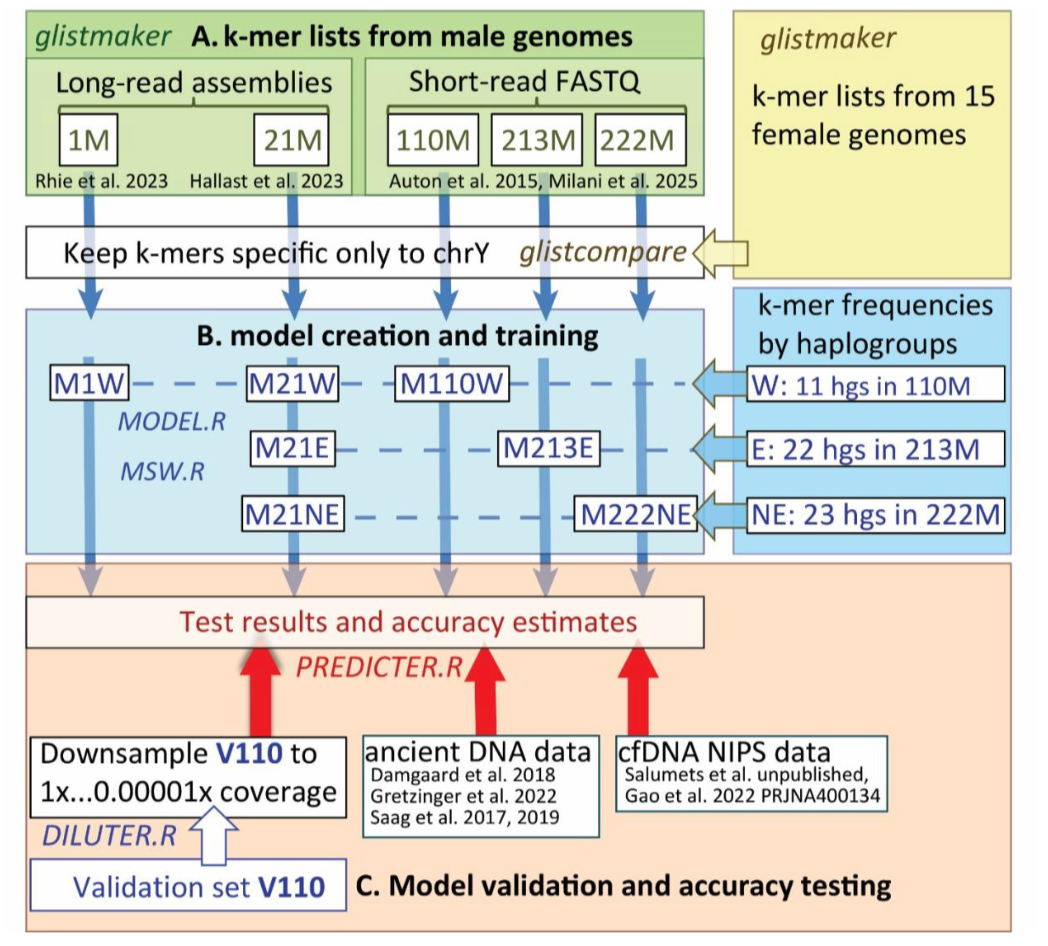
图2. 从k-mer列表生成的Y染色体单倍群预测模型及其在验证集、古DNA和NIPT数据上的测试。这里展示的工作流程包含三个阶段,用于生成k-mer列表和模型的R脚本和GenomeTester4命令名称以斜体显示在相关任务旁边:(A) 用于生成k-mer列表的chrY来源选自高覆盖度(>20X)长读长或短读长测序数据。1M、21M、110M、213M和222M分别指1、21、110、213和222个男性基因组的来源。对于1M,使用了Y染色体的三个独特区域(表S1)进行k-mer选择。对于其他来源,使用了整个Y染色体。使用glistmaker从每个来源生成k-mer列表。使用glistcompare识别并移除也映射到女性基因组的k-mers(表S6)。(B) 为单倍群预测定义了三个具有全球和局部单倍群分布的训练集(表S4),并在每个训练集中使用MWS.R识别10K到100K个单倍群特异性k-mers。使用MODEL.R通过训练集训练具有特定k-mer数量和单倍群选择的模型。(C) 使用PREDICTER.R在验证集V110、1000 GP、HGDP、SGDP、古DNA和NIPT数据上进行模型准确性测试。
模型验证
我们首先在从1000 GP、HGDP和EstBB数据中选择的110个个体的验证集(V110)上测试了单倍群预测模型的性能,这些个体均未用于模型训练(表S7)。验证集中每个个体的单倍群都已通过SNV数据确定。验证集包括代表全球人类Y染色体多样性的11个基本单倍群(AB、C、E、G、H、IJ、LT、N、O、Q和R)中各10个个体。验证集中的每个个体都被降采样到10个不同的覆盖度值,范围从1x到0.00001×,以测试该方法的极限。
首先,为了明确需要多少不同的Y染色体组装作为k-mer列表的来源,以便从ulcWGS数据准确确定Y染色体单倍群,我们比较了M1W、M21W和M110W模型在验证集上的表现(图3,表S8,S9)。我们发现M21W和M110W模型在1x到0.001x覆盖度范围内都提供了高准确度(>0.95),而仅使用单个Y染色体组装作为来源在所有覆盖度下表现都较差,在1×覆盖度时最高准确度仅为0.65。M110W模型在0.0005×到0.005×覆盖度范围内显示出比M21W更高的准确度,而在更高覆盖度范围内,差异并不显著。
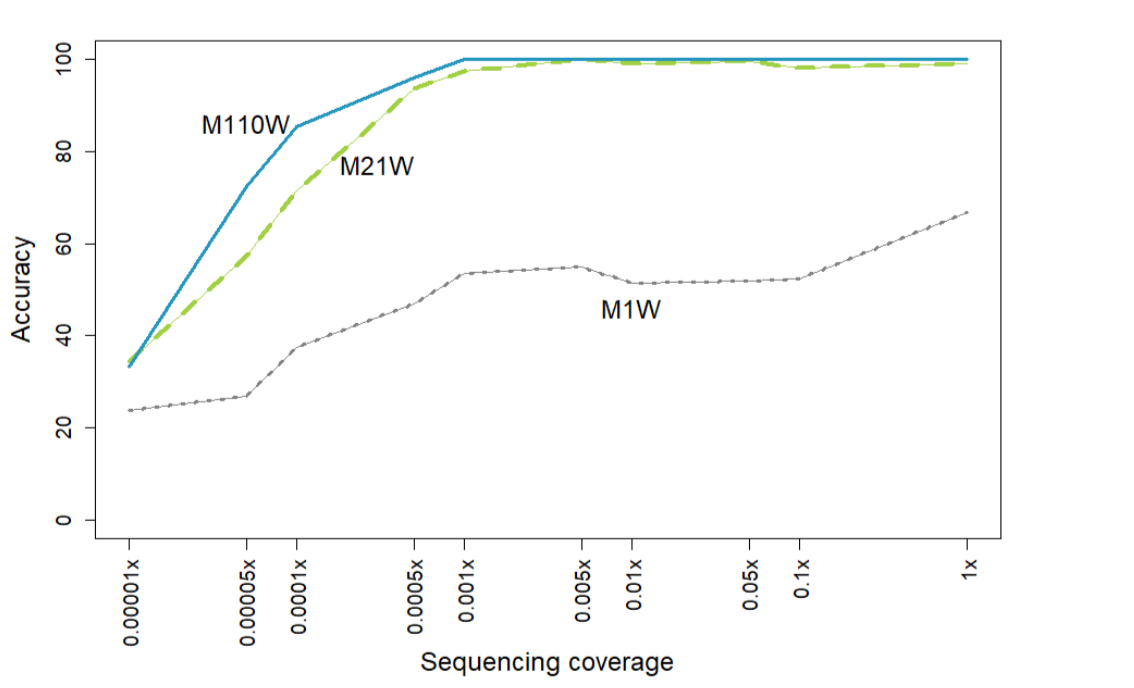
图3. 低覆盖度范围内的单倍群预测准确度。
展示了三个预测模型,它们基于单个染色体三个重复区域(1Y)和多个(21Y, 110Y) chrY组装中选择的k-mer。这三个模型中,每个模型都使用了50000个k-mers来预测验证集中110个个体的单倍群,这些个体代表了全球分布的11个基本单倍群。
接下来,为了确定准确预测chrY单倍群需要多少k-mers,我们比较了基于从每个单倍群选择的10K到100K k-mers的模型性能。在每个测试模型中,我们使用21Y(21个Y染色体的长读长组装,Hallast等,2023)作为k-mer列表的来源。我们发现在覆盖度等于或高于0.0005x时,所有模型都表现出很高的准确度(>0.95)(图4)。基于10K k-mers的模型在低于0.0005x的覆盖度范围内准确度明显较低。在最低覆盖度0.00001x时,20K模型的表现也较差,但我们在测试的覆盖度范围内,没有观察到基于30K或更多k-mers的模型之间存在重大性能差异。
我们通过对来自单倍群N3的一个芬兰个体的降采样复制品测试了污染对Y-mer单倍群预测准确度的影响。我们向这个测试样本中添加了来自另一个单倍群R1a个体的reads,同时将总reads数保持在0.01x覆盖度不变。我们使用Y-mer确定了这些混合个体reads的单倍群,结果表明Y-mer的单倍群预测在污染率高达30%时仍然保持高度稳健(图5)。
图4. 每个单倍群选择不同数量k-mers的影响。使用
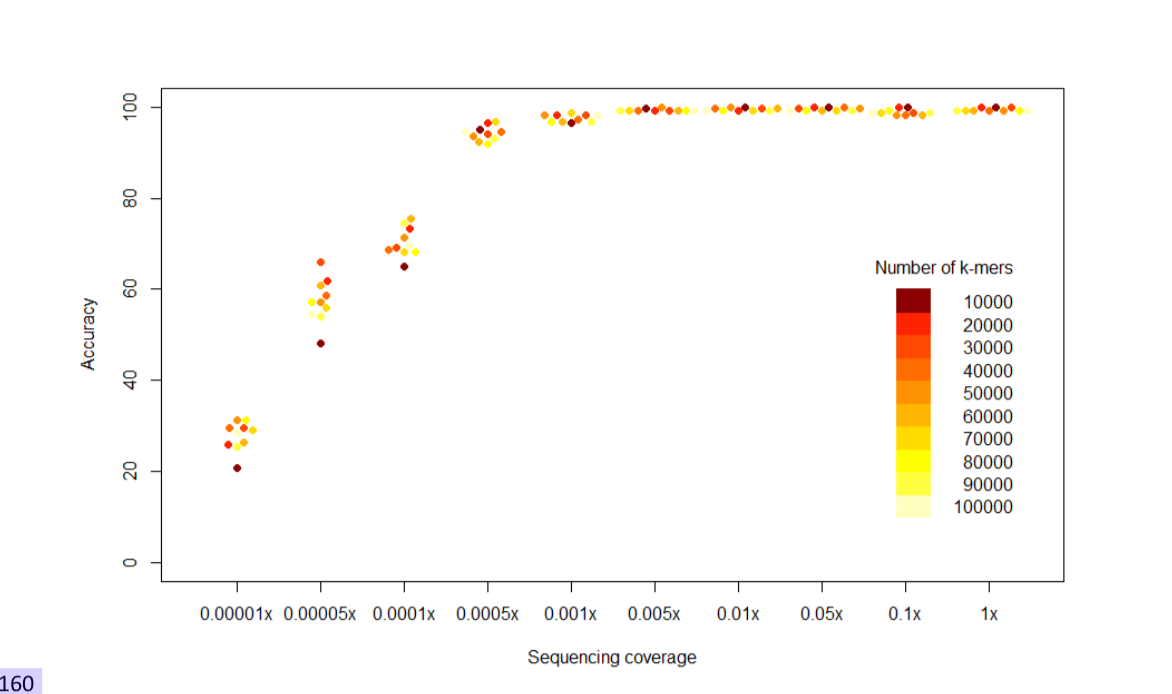
图4. 每个单倍群选择不同数量k-mers的影响。使用M21W模型说明;在V110验证集稀释样本上的单倍群预测准确度。最后,我们还在HGDP(表S11)和SGDP(表S12)数据中的个体上测试了在1000 GP数据上训练的模型(表S10)。表S13总结了在1000 GP、HGDP和SGDP数据集中的预测准确度估计,显示W和E模型在不同数据集中均保持一致的高准确度(>0.95)。E模型在HGDP中略低的准确度(0.9-0.95)主要是由于对R1b13-FT289648和N3a2d-M1932谱系的错误分配,这些谱系在HGDP数据中有多个个体独特存在,而在1000 GP数据中缺失,因此未在模型中得到表示。
古DNA数据的模型测试
我们在V110验证集上的单倍群预测准确度测试包括来自与训练集相同的现代人群的个体。为了探索我们的模型在单倍群组成与现代训练集不同的数据集上的表现,我们转向了古DNA数据。
我们首先在来自欧亚草原带的91个男性个体的古基因组数据上测试了单倍群预测模型,这些样本距今1500-4500年,全基因组测序深度范围为0.029-8.7x(Damgaard等,2018)。除了我们的现代参考和Damgaard数据之间的时间差异外,我们用于模型训练的数据中没有包括任何现代草原带或中亚人群。Damgaard等人2018年报告了47个个体的基本Y染色体单倍群,而相当大比例的44个男性样本没有单倍群分配,这可能是由于其低覆盖度。为了生成用于模型测试的预期单倍群列表,我们调用了256,463个二元单倍群信息SNV,并按照Hui等人2024年描述的方法确定了Damgaard等人数据中所有91个草原带个体最可能的chrY单倍群(表S14)。我们对44个具有先前分配的个体所做的所有单倍群分配在基本单倍群水平上都与Damgaard等人2018年所做的分配相匹配。在这个古代数据集中检测到的一些单倍群,如C3、I3、N5、O6a、Q1c、Q1g、R1b13和R1b16,在现代数据集中要么极其罕见,要么未包含在我们的训练集中。
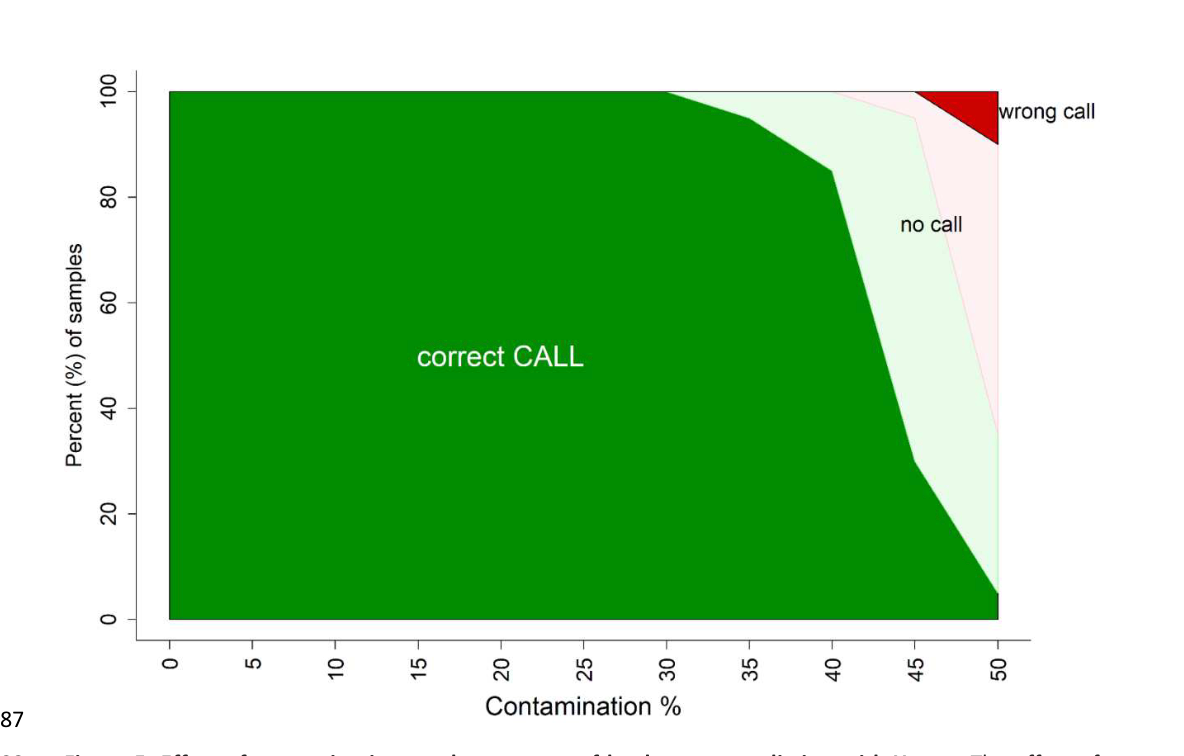
图5. Y-mer单倍群预测准确度的污染影响。通过将单个供体(HG03687,单倍群R1a)的reads以5%的增量(在x轴上显示)添加到受体样本(HG00280,单倍群N3)的降采样复制品中,同时将总覆盖度保持在0.01x不变来估计污染的影响。具有不确定单倍群(p值 >= 0.05)的预测标记为"无法判定"。不确定但正确的单倍群用浅绿色表示,不确定的错误单倍群预测用粉色表示。
为了测试Y-mer在系统发育多样性的古代草原带数据上的表现,我们使用6种不同的基于k-mer的模型预测了所有91个个体的chrY单倍群(表S14)。在使用21个Y染色体T2T组装衍生的k-mer列表(模型M21W)预测的11个最基本单倍群水平上,我们观察到SNV和k-mer数据之间的单倍群判定有很高的匹配率(94%)(表1)。在训练用于预测更高分辨率单倍群的模型(M21E、M222E、M21NE、M222NE)中,我们观察到72-86%的准确度,其中特别是在未用于模型训练的单倍群(如M21NE和M222NE模型中的C和O单倍群)中出现更多不匹配。
在M222NE模型中,所有78个SNV确定的单倍群包含在模型中的个体都被正确预测。在13个SNV数据预期的单倍群未用于我们模型训练的案例中,6个案例的模型预测是系统发育上密切相关的谱系(如I2代替I3,R1b3代替R1b16),7个案例是单倍群LT,这在系统发育上比训练集中最接近的单倍群更远。错误的单倍群预测包括值得注意的是,以高置信度(p<0.05)做出的单倍群LT分配,而只有少部分错误预测是由于低置信度判定造成的。由于对p值进行过滤似乎并未提高单倍群预测的准确度,我们考虑了不同模型做出的单倍群预测的一致性。我们发现对于45个个体,所有6个模型的单倍群分配都是一致的,且这些预测始终是正确的。值得注意的是,正确预测包括覆盖度最低的个体。使用p<0.05的阈值进行单倍群预测仅略微改善了准确度(表1)。
表1. 三个古DNA数据集上单倍群分配模型的准确度(表S14-16)
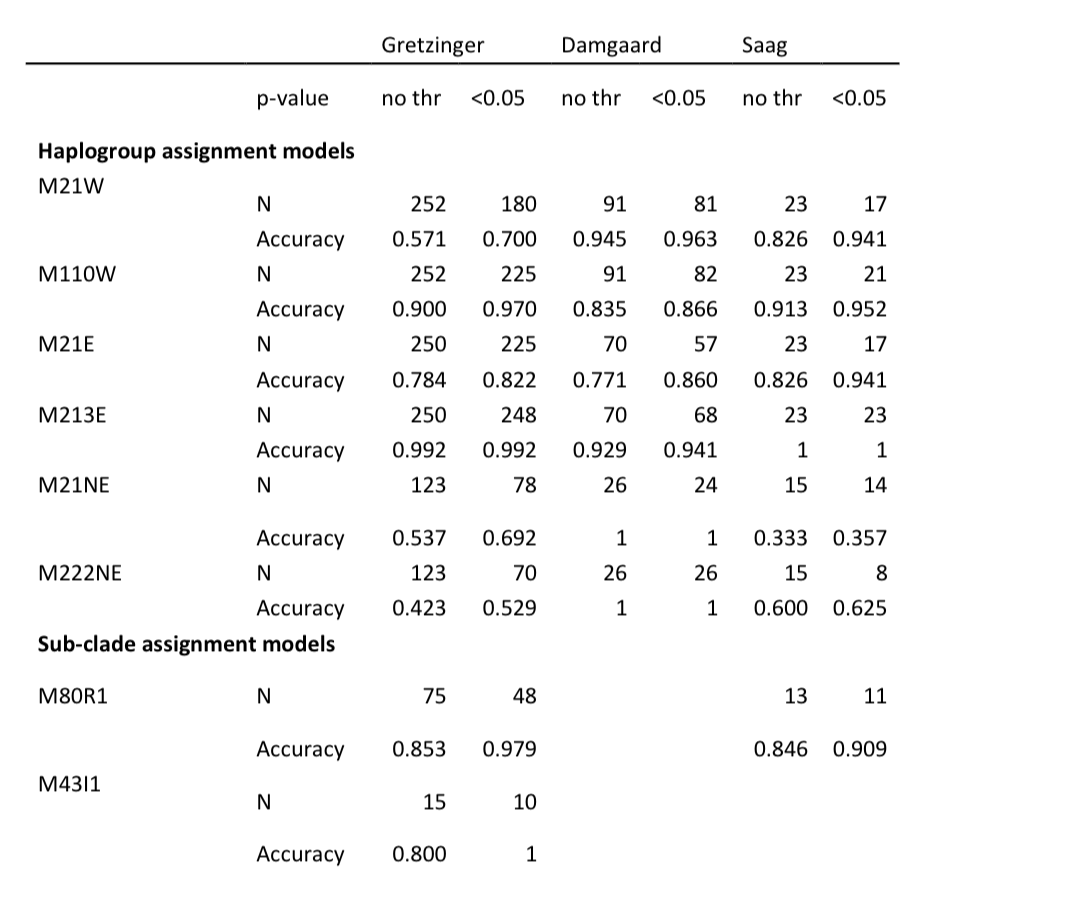
注:N - 测试中考虑的个体数量,'no thr' - 未使用p值阈值;'<0.05' - 仅保留p值<0.05支持的单倍群分配。Gretzinger数据(Gretzinger等,2022);Damgaard数据(Damgaard等,2018);Saag数据(Saag等,2017,2019)。
为了测试高分辨率模型M21E、M213E、M21NE和M222NE在单倍群组成与我们训练集更相似的数据上的准确性,我们转向了另外两个古DNA数据集,一个来自早期中世纪西北欧(Gretzinger等,2022),另一个来自青铜时代和铁器时代的爱沙尼亚(Saag等,2017,2019)。这些数据集包括了与我们训练集在时间上有差异的个体,但在亚支系水平上仍具有相似的chrY单倍群组成(表S14-16)。与Damgaard等人的数据相比,M21E、M21NE和M222NE模型的性能改善有限或没有改善(表1)。然而,M213E模型在Gretzinger等人和Saag等人的数据集中都显示出较高(>0.98)的基本单倍群分配准确度。由于M213E模型与M222NE的区别在于它不区分最近分化的R1b和I1亚支系,值得注意的是,在分别对被分配为R1和I1支系的个体应用M80R1和M43I1模型后的单倍群预测准确度明显更高(>0.8),而在M222NE模型中(<0.53),这些亚支系分配是在全球chrY系统发育的更高层次单倍群分配的背景下进行的。
在无创产前筛查(NIPS)数据上的模型测试
另一个传统基于SNV方法难以进行Y染色体预测的低覆盖度数据来源是无创产前筛查(NIPS)数据。这些数据来自于孕妇血液中的循环游离DNA(cfDNA)样本,广泛用于识别胎儿发育各阶段的染色体结构异常。我们使用来自中国(Xu等,2018)和爱沙尼亚(Žilina等,2019)的NIPS数据,并关注已独立确定胎儿性别为男性的病例,结果表明Y-mer能够从爱沙尼亚样本0.006-0.12x的测序深度(表S17)和中国样本0.0009-0.009x的测序深度(表S18)中高置信度地预测胎儿单倍群。
虽然我们没有个别NIPS样本的参考数据来估计Y-mer的准确性,但中国和爱沙尼亚队列中的单倍群预测结果在大多数单倍群上都与独立采样队列得出的中国和爱沙尼亚频率预期相匹配(表S19)。在中国NIPS单倍群预测中,我们观察到预测频率和观察频率总体匹配良好,但C单倍群频率略高于预期,O3(ISOGG命名法中的O2a2)频率低于预期,这可能反映了NIPS和1000 GP数据在招募中国南方和北方个体时的差异,因为这些单倍群在这些地区显示相反的频率梯度(Li等,2023)。
讨论
从法医环境或考古环境中保存不佳的古代人类遗骸中提取的DNA痕迹来确定个体祖先通常具有挑战性,这是由于样本中宿主DNA分子数量少、片段化和死后损伤等原因造成的。由于Y染色体缺乏重组,其变异随时间沿着简单的系统发育树积累。即使从低覆盖度数据中也可以稳健地推断出这棵树的主要分支,这是因为积累变异具有层级冗余性。此外,与线粒体和常染色体多样性相比,现代人群中观察到的Y染色体区域分化程度更高(Karmin等,2015),这使得Y染色体单倍群预测在遗传祖先推断中具有吸引力。在本研究中,我们证明了可以使用确定男性特异性k-mers相对丰度的方法,从ulcWGS数据中准确和有效地预测人类Y染色体单倍群。
单倍群预测的准确性、分辨率和稳健性,无论是使用SNV还是本文描述的Y-mer方法,除了样本质量和测序覆盖度外,还取决于参考面板的大小和多样性以及所使用的信息变异数量。我们使用全球和欧洲训练集和验证集进行的测试表明,仅使用单个T2T Y染色体参考基因组作为k-mer提取源的模型表现不佳,在测试的降采样验证数据覆盖度范围内准确度低于80%。使用从21、110和213个不同Y染色体来源提取的k-mers的模型表现更好,在覆盖度范围>0.001x时准确度高于95%(图3)。由于我们没有观察到M21W与M110W以及M21E与M213E模型在性能上的重大差异,我们可以得出结论,超过20个具有系统发育多样性的Y染色体面板足以作为基础单倍群判定的k-mer来源。
我们证明了使用从系统发育上不同单倍群的个体训练的模型(M21E)时,Y-mer的单倍群预测对污染具有鲁棒性(图5)。然而,使用区域特异性单倍群集训练的模型(M21NE),包括仅在过去5000年内分离的I1和R1b亚支系(Karmin等,2015),即使在没有污染存在的情况下表现也较差(表1)。这种性能下降可能是由于为给定亚支系提取的k-mer列表中重叠k-mer比例越来越高造成的。这一结果强调了在未来开发详细亚支系预测模型时需要使用额外的过滤器来消除k-mer重叠,或者在我们当前的方法中,需要使用多样化且平衡的训练集。
虽然我们没有看到M21W和M110W模型之间的改进(图3),但对于需要更高单倍群分辨率的模型,可能还需要调整用于提取k-mer的Y染色体来源数量。我们的分析表明,为了进行稳健的单倍群预测,用于区分模型中包含的每个单倍群的k-mer数量必须足够高(>10,000)。然而,我们对具有越来越多k-mer数量的模型的比较显示,在超过20,000个单倍群特异性k-mer的模型中没有检测到准确度的增加(图4),这表明对于计算效率而言,具有20,000-50,000个k-mer的模型可能代表最优解决方案。
当对验证集使用Y-mer时,如果验证集的单倍群组成与训练集不同,我们观察到更高的不匹配率,特别是在罕见亚支系方面。在不同单倍群水平上训练的模型支持的预测大多是正确的,这表明在相同数据上应用多个模型可以帮助区分那些被多个模型稳健支持的预测和那些仅由个别预测支持的低置信度预测。
在草原带古DNA数据的案例中进一步说明了在相同数据上应用多个模型的必要性。我们的基础单倍群预测模型M21W以高准确度(~95%)进行单倍群判定,而针对特定区域单倍群组成的预测模型(根据欧洲或更具体地说某些模型基于东北欧数据调整)显示出更多的不匹配,特别是在未包含在模型中的单倍群方面。
虽然我们发现被多个模型支持的单倍群预测大多是正确的,但这个案例研究强调了在古DNA研究中未来使用Y-mer工具时,需要谨慎选择具有适当单倍群组成的模型和训练集。建议通过对高覆盖度个体样本进行SNV分析,初步了解古代群体的单倍群组成,以指导这些模型。在可获得此类高覆盖度数据的情况下,它可以显著提高Y-mer从较低覆盖度范围数据预测单倍群的准确性。根据我们的测试,当验证集被降采样到较低覆盖度时(图3),人类chrY 0.001x测序深度似乎足以进行准确的单倍群预测。
在更广泛的全球范围内进行单倍群判定时,建议采用分层推断策略。首先可以应用覆盖所有主要单倍群的通用模型(如M21W)来确定主要单倍群,然后再应用特定的子单倍群模型进行进一步推断。我们通过R1和I1单倍群模型展示了这种方法。对欧洲两个古DNA数据集(Saag等,2017, 2019; Gretzinger等,2022)的分析显示,使用M213E模型进行基础单倍群预测具有很高的准确性,该模型不区分R1b和I1最近分化的子支系。M222NE模型旨在预测这些子支系,其训练集将这些子支系与全球范围的单倍群多样性相结合,表现较差(准确度<0.53)。相比之下,当使用M110W或M213E模型预测的R1或I1单倍群进一步用子支系特异性模型解析时,对于高置信度(p<0.05)的预测显示出很高的准确度(>0.95)。这些结果表明,在单倍群预测中采用两阶段策略可能更可取,即先判定基础单倍群,再单独进行子支系判定,而不是使用结合不同层次单倍群多样性的模型。
我们对中国和爱沙尼亚NIPS数据的分析进一步证实了Y-mer在低覆盖度范围(0.001-0.12x)的男性胎儿Y染色体数据上表现良好,我们获得的单倍群频率分布与相关参考数据的预期相似。虽然这些结果表明欧洲和亚洲最常见的单倍群可以以足够的准确度预测,但我们观察到在子支系水平上进行单倍群区分的模型准确度下降,这进一步强调了我们目前描述的方法在需要高置信度和分辨率的目的(如确定遗传关系)方面的局限性。然而,在已经通过其他方法(如通过同源性或状态同一性方法)独立确定遗传关系的情况下(Monroy Kuhn, Jakobsson和Günther, 2018; Popli, Peyrégne和Peter, 2023),Y-mer分析可用于测试(排除)父系关系的可能性,即使承认在通用Y染色体单倍群水平上的匹配不能构成父系关系的证明。
总之,我们提出了一个新的基于k-mer的工具Y-mer,用于预测Y染色体单倍群。我们表明Y-mer能够从超低覆盖度(>0.1x)数据中准确预测基础chrY单倍群。因此,在需要获取个体祖先的基本、低分辨率信息,但由于成本或样本数量不足等原因无法或不实际进行高覆盖度测序的情况下,这种方法非常有用。为此,我们提供了已经测试过的工具和模型,以及开发新的更具体模型的指导(https://github.com/bioinfo-ut/Y-mer/)。
对于古DNA研究或法医案例分析,这种方法可以潜在地使更多个体样本可用于Y染色体祖先分析,当部分样本已有高覆盖度/高质量数据时,这种分析可以提供更多信息。我们表明,当Y-mer的模型使用与目标群体单倍群组成匹配的数据进行训练时,其表现更准确,这突显了在需要详细的子单倍群水平区分的情况下需要采用定制方法。
除了在Y染色体数据方面的潜在应用外,这里描述的基于k-mer的方法也可能扩展到常染色体的祖先分析。考虑到在使用长读长序列组装的着丝粒区域检测到的高遗传变异率以及着丝粒周边区域的低重组率(Logsdon等,2024),研究来自(着丝粒周边)单倍型的k-mer可能为从低覆盖度数据进行常染色体祖先扫描提供新的前景,尽管考虑到常染色体和Y染色体DNA遗传方式的差异,这与这里描述的Y染色体分析并不完全相同。另外,可以筛选和使用常染色体群体特异性k-mer丰度峰值的全基因组扫描来进行低覆盖度数据的祖先映射。开发此类工具需要更大的具有祖先多样性的参考面板,如目前正在开发并可能在不久的将来可用的基于图的泛基因组。
方法
人类Y染色体命名法
大多数之前的群体遗传学研究在进行祖先分析和Y染色体单倍群判定时,仅使用了Y染色体的X-退化区域,这些区域占Y染色体总长度不到五分之一,被认为适合短读长序列比对(Francalacci等,2013; Mendez等,2013; Poznik等,2013, 2016; Wei等,2013; Hallast等,2015; Karmin等,2015)。
人类Y染色体单倍群是通过独特的等位变异组合(通常是SNV)来定义的,这些变异在共享父系祖先的个体中共同出现,而在其他被检测的个体中不存在。自2002年基于245个标记在全球代表性样本中建立Y染色体单倍群命名系统以来(Consortium, 2002),已经有多次更新尝试。
这个字母数字命名系统从2005年到2020年一直由一大群公民科学家进行更新(https://isogg.org/tree/)。由于序列数据量的不断增加,一些更新后的ISOGG单倍群标签超过20个字符,而Y系统发育树的许多亚支系仍然标记不完善。为了找到更简短和更稳定的替代方案,van Oven等人(van Oven等,2013)提出了一个最小参考树,而Karmin等(2015)提出了一个更短的、基于时间深度约束的单倍群标记系统。本研究在提及单倍群名称时将使用后者系统。在数据来源使用了平行系统(如基于ISOGG的标记)的情况下,为了清晰起见,将明确提及这些系统。
 陕公网安备61011302002223号
陕公网安备61011302002223号