印度 50,000 年进化历史:对健康和疾病变异的影响
印度在基因组调查中一直代表性不足。我们从印度的2,762个个体中生成了全基因组序列,捕捉了大多数地理区域、语言群体和历史上代表性不足的社区的遗传多样性。我们发现大多数印度人主要携带来自三个祖先群体的血统:南亚狩猎采集者、欧亚草原牧民以及与伊朗和中亚文化相关的新石器时代农民。个体间广泛的纯合性和相同祖先共享反映了由于最近向内婚制的转变而导致的强烈创始者效应。 我们发现印度人的大部分遗传变异源于约50,000年前发生的一次主要走出非洲的迁徙,随后有1%-2%的基因流来自尼安德特人和丹尼索瓦人。值得注意的是,印度人在全球群体中展示了最大的变异,并拥有最高数量的特定人群尼安德特人祖先片段。最后,我们
50,000 years of evolutionary history of India: Impact on health and disease variation
亮点
- 来自约2,700个全基因组序列的印度遗传变异洞察
- 鉴定印度伊朗农民相关祖先的来源
- 印度尼安德特人和丹尼索瓦人祖先的特征分析
- 发现印度特定人群和疾病易感性变异
摘要
印度在基因组调查中一直代表性不足。我们从印度的2,762个个体中生成了全基因组序列,捕捉了大多数地理区域、语言群体和历史上代表性不足的社区的遗传多样性。我们发现大多数印度人主要携带来自三个祖先群体的血统:南亚狩猎采集者、欧亚草原牧民以及与伊朗和中亚文化相关的新石器时代农民。个体间广泛的纯合性和相同祖先共享反映了由于最近向内婚制的转变而导致的强烈创始者效应。
我们发现印度人的大部分遗传变异源于约50,000年前发生的一次主要走出非洲的迁徙,随后有1%-2%的基因流来自尼安德特人和丹尼索瓦人。值得注意的是,印度人在全球群体中展示了最大的变异,并拥有最高数量的特定人群尼安德特人祖先片段。最后,我们讨论了这种复杂的进化历史如何塑造了次大陆上的功能和疾病变异。
关键词
简介
印度拥有超过14亿人口和约5,000个人类学上明确定义的民族语言和宗教社区,是一个具有非凡多样性的地区。1 然而,印度人口在基因组研究中仍然代表性不足。近期的测序研究,如千人基因组计划(1000G)2、英国生物银行3、TopMed4、西蒙斯基因组多样性小组5、人类基因组多样性小组(HGDP)6和亚洲基因组计划7,8都纳入了印度人群。然而,除了亚洲基因组计划7,8外,这些研究要么仅包含很少的个体,要么主要采样了印度境外的侨民社区,导致对印度遗传变异的代表性有限(且有偏见)。
因此,关于印度人口历史的许多开放性问题仍未得到解答:人类何时首次从非洲迁移到印度——是作为主要走出非洲迁徙的一部分,还是早期沿南部沿海迁徙路线?9,10 古人类如尼安德特人和丹尼索瓦人对印度人群的基因流动的贡献和遗留是什么?新石器时代农业和语言传播等最近的技术创新如何影响了印度的遗传变异和疾病?
理解人口历史——过去的迁徙、人口瓶颈和混合事件——对追踪人口起源和有效疾病映射的基础都很有用。最近的研究表明,对人口结构考虑不足可能会导致全基因组关联研究中出现假阳性结果。11,12 相反,利用人口历史可以增强检测真实关联的能力,并提供关于致病变异的来源和随时间变化动态的见解。13,14 例如,来自古代人群的基因流动对人类适应和适应度产生了重大影响,影响了从高海拔适应到糖尿病和传染病风险的众多特征。15,16,17 因此,全面了解人口历史是有效疾病映射的重要第一步。
为了获得印度遗传多样性的详细图景,我们生成了2,762个个体的深度覆盖基因组序列。我们的数据是印度长期老龄化研究-痴呆症诊断评估(LASI-DAD)18的一部分,这是一项针对60岁及以上个体的基于人群的前瞻性基因组队列研究。LASI-DAD包含来自印度18个不同州和联邦直辖区的具有全国代表性的个体数据(图1A),每个州的样本量中位数为157个个体(参见STAR方法和数据S1,第1部分)。它包括多样化的地理区域(包括农村和城市地区)、各种语言家族的使用者(如印欧语系、达罗毗荼语系和藏缅语系)以及历史上代表性不足的社区(如指定部落[ST]、指定种姓[SC]和其他落后阶层[OBC]),提供了印度遗传变异最全面的快照。
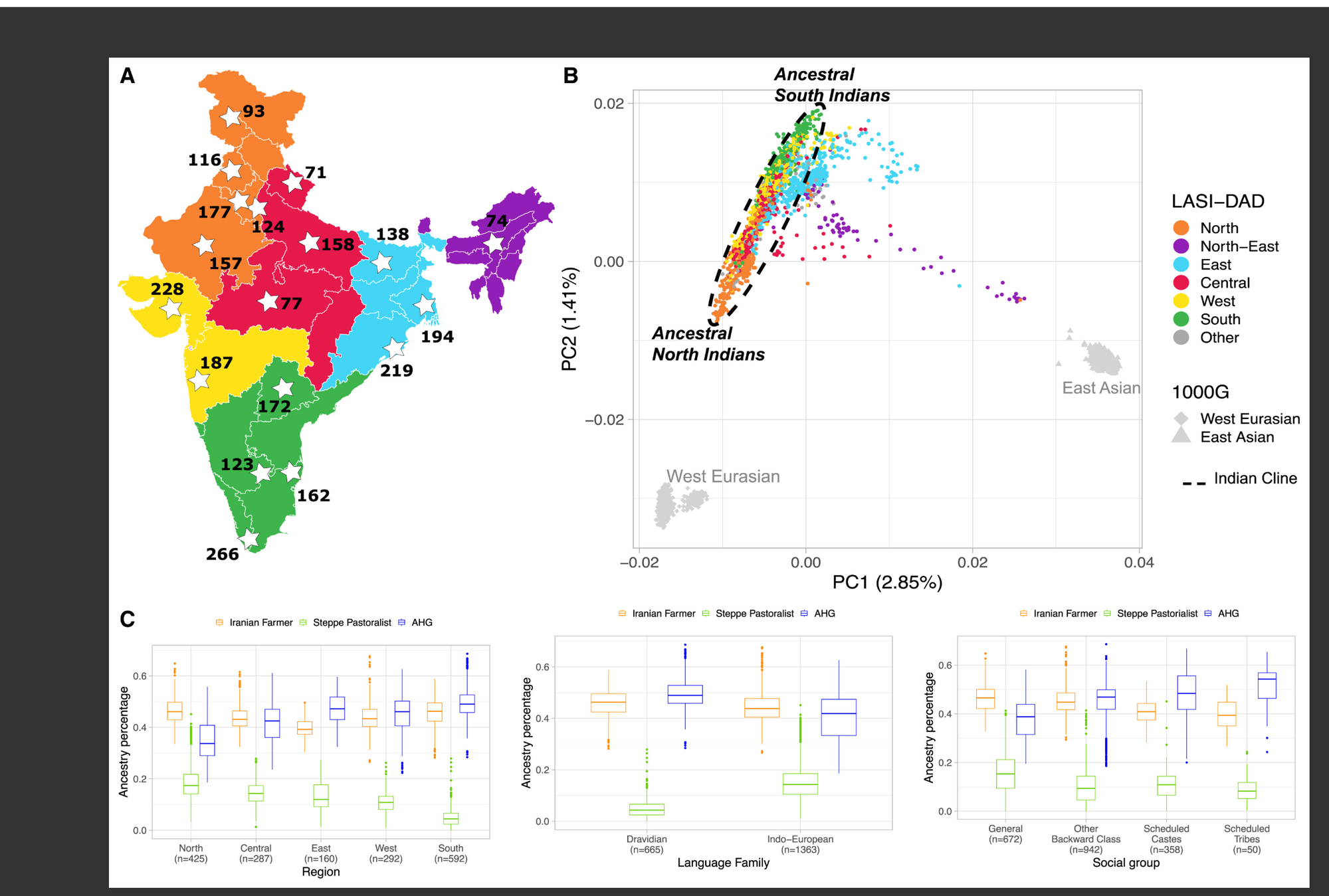
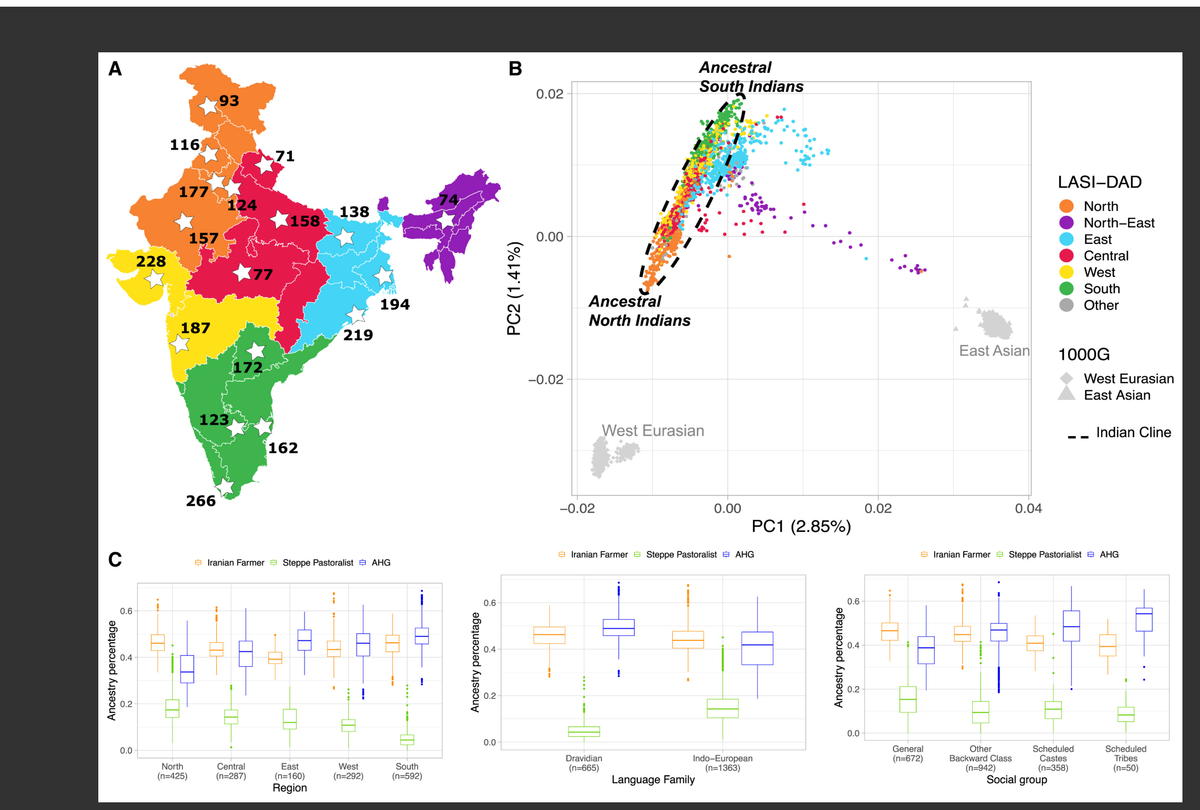
图1 印度的人口结构和混合
(A) LASI-DAD个体在印度的采样位置,按区域(北部、东北部、中部、南部、东部和西部)着色用于分析。
(B) 对LASI-DAD中的印度人和1000G中的西欧亚(EUR)、东亚(EAS)和南亚(SAS)血统个体进行的主成分分析(PCA)。我们展示了前两个主成分的投影,按出生地区着色。我们在HGDP的西欧亚和东亚人群中发现类似的结果(Data S1,图S4.5)。
(C) 使用Sarazm_EN(橙色)作为伊朗农民相关祖先的代表,Central_Steppe_MLBA(绿色)作为草原牧民相关祖先的代表,以及AHG(Onge)(蓝色)作为AASI相关祖先的代表,对印度梯度上每个个体的祖先比例进行分析。祖先比例按每个个体的区域(左)、语言家族(中)和社会群体(右)进行比较。箱形图的盒边界表示第25和第75百分位数;中心线表示中位数;须表示数据中的最小/最大值或第25/75百分位数+1.5×四分位距。
结果
数据和遗传变异目录
总共有2,762名LASI-DAD参与者,包括22个三联体(母亲-父亲-孩子),在MedGenome(印度班加罗尔)进行了平均读取深度为30×的测序。原始全基因组序列被发送到宾夕法尼亚大学的阿尔茨海默病基因组中心(GCAD)进行联合调用和质量控制(参见STAR方法;数据S1,第2部分)。共有2,679个个体和7320万个常染色体双等位基因变异通过了质量控制筛选,包括6710万个单核苷酸变异(SNVs)和604万个插入-缺失(indels)。
我们确定了2400万个SNVs和220万个indels在现有人类基因组变异数据库中不存在,如千人基因组计划和基因组聚合数据库(gnomAD)19,这突显了这些数据库在代表多样化人群方面的局限性。绝大多数(>99%)这些变异是罕见的(频率低于1%),其中68%是单例变异(数据S1,表S2.1)。基因组分型是使用SHAPEIT420(不使用参考面板)进行的,我们在三联体中估计的低相位切换错误率不到1.13%(数据S1,表S3.1)。
我们的数据集代表了印度的人口多样性。它包括出生于23个州和联邦直辖区的个体,来自农村(63%)和城市(37%)地区。数据集涵盖约26种语言的使用者,其中74%的个体报告为印欧语系使用者,25%为达罗毗荼语系使用者。它包括来自不同社区的个体,包括部落(ST: 4%)和种姓(SC: 18%和OBC: 44%)群体。21,22 招募的男性和女性人数几乎相等,女性占我们数据集的52%。对于许多分析,我们根据个体的出生地将其分为六个主要地理区域:北部(n = 555)、西部(n = 385)、中部(n = 373)、南部(n = 715)、东北部(n = 73)和东部(n = 530)(数据S1,第1部分)。在进行质量控制检查并排除一级亲属后,我们使用了2,620个个体的样本进行以下大多数分析,除非另有说明(数据S1,第2部分)。
人口结构和混合
为了研究印度人与全球其他人群的关系,我们将LASI-DAD与1000G23和HGDP6数据集结合起来,进行了主成分分析(PCA)24和ADMIXTURE分析25。与先前的报告一致26,27,我们发现印度的人口结构与西欧亚相关(1000G EUR)和东亚相关(1000G EAS)血统的个体有关(图1B和数据S1,图S4.1)。当包括来自HGDP的中亚和中东人群时,我们获得了质量上相似的结果,这强调了PCA中观察到的模式反映了西欧亚相关性而非仅仅是欧洲相关性(数据S1,图S4.5)。
与之前主要对印度隔离的内婚社区进行采样的研究不同26,28,29,我们使用了基于网格或人口密度的采样方法。值得注意的是,我们沿PC1和PC2观察到祖先成分的更连续变异。PCA中有三个主要集群——一个包括印度北部和南部大多数个体的集群,这些个体表现出与西欧亚人不同程度的关联,被称为"印度梯度"(图1B和数据S1,图S4.2和S4.3)。先前已经证明印度梯度反映了来自两个祖先群体的不同比例血统:携带大比例与西欧亚人相关血统的"祖先北印度人"(ANI),以及与西欧亚人仅有远缘关系的"祖先南印度人"(ASI)26,27。
最近的古DNA分析表明,与安达曼狩猎采集者(AHG)有远缘关系的(未采样的)南亚原住民("古代祖先南印度人"[AASI])与古代伊朗农民混合形成了ASI,而ASI又与古代欧亚草原牧民相关群体混合形成了ANI。因此,ASI和ANI都是混合群体30(框1)。
| AASI | Ancient Ancestral South Indians represent an Indigenous, unsampled South Asian population that is one of the most ancient lineages in South Asia |
|---|---|
| ASI | Ancestral South Indians represent a hypothetical group that has ancestry related to AASI and ancient Iranian farmers |
| ANI | Ancestral North Indians represent a hypothetical group that has ancestry related to ASI and Eurasian Steppe pastoralists from CentralSteppeMLBA |
| AHG | AHG refers to present-day Indigenous Andaman Islanders, the Onge, who are related to the unsampled Indigenous South Asians30 (n = 15) |
| Indus Periphery Cline | Indus Periphery Cline is a heterogeneous group of 11 outlier samples from Bronze Age cultures of Shahr-i-Sokhta and Bactria Margiana Archaeological Complex that has been shown to have Iranian farmer-related and some AHG-related ancestry in an earlier study.30 Indus Periphery West (I8726) is a single individual with the highest Iranian farmer-related ancestry among the Indus Periphery Cline, dated to ∼3,100–3,000 BCE |
| Central_Steppe_MLBA | The individuals from the Central Steppe Middle to late Bronze Age are considered as the source for Yamnaya Steppe pastoralist-derived ancestry in South Asia30 (n = 34) dated to ∼2,000–900 BCE |
| Sarazm_EN | 4th millennium BCE farmers and herders from Sarazm, Tajikistan, dated to ∼3,600–3,500 BCE (n = 2) |
| Parkhai_Anau_EN | Eneolithic individuals from Tepe Anau and Parkhai in Turkmenistan dated to ∼3,500–2,900 BCE (n = 9) |
| Namazga_CA | Chalcolithic individuals from Namazga in Turkmenistan dated to ∼3,300–3,200 BCE (n = 2) |
在印度梯度之外,我们发现了两个主要的个体群集(n = 494):一个群集靠近梯度的ASI端,另一个位于中心位置之外但表现出与东亚相关群体(1000G EAS)的明显关联(图1B)。前者主要包括来自印度中部和东部的个体,其中大多数来自奥里萨邦,那里主要讲印欧语系和南亚语系语言。第二个群集包括来自印度东部和东北部地区的个体。西孟加拉邦是这个群集中最具代表性的邦,个体约有∼5%的祖先与东亚人相关(数据S1,第4部分)。通过使用ALDER31测量混合相关的连锁不平衡,我们推断出东亚相关的基因流发生在50代前或约公元520年(数据S1,图S4.15),这与早期对孟加拉国个体研究的估计一致32。这个时间与笈多帝国的崩溃相吻合,尽管一些混合也可能随着水稻种植从东亚传播而更早发生33,34。与东亚相关的群集还包括来自阿萨姆邦的说藏缅语的个体。PCA显示这一群体中的个体在与东亚的关联性上存在显著的异质性,表明最近有基因流动(图1B)。我们的ADMIXTURE25分析反映了在PCA中观察到的模式(数据S1,图S4.8)。
为了模拟印度的祖先构成,我们使用了qpAdm,该方法比较了目标人群与一组参考和外群人群之间的等位基因频率相关性。35,36 首先,我们检验了由古代伊朗农民相关、欧亚草原牧民相关和AHG相关群体组成的三路模型如何解释印度梯度上个体的遗传变异(图1B)。根据Narasimhan等人的研究,30 我们使用印度外围西部(作为印度外围梯度的一部分——一个由沙赫尔-索赫塔和巴克特里亚马尔吉亚那考古复合体青铜时代文化的11个异常样本组成的异质群体)作为伊朗农民相关祖先的代表,中亚草原中晚期青铜时代(Central_Steppe_MLBA)作为草原牧民相关祖先的来源,以及AHG相关个体来代表AASI祖先30(框1)。我们发现这个三路模型能够很好地解释印度梯度上大多数(>90%)个体的遗传构成,只有少数例外(我们将"良好拟合"定义为qpAdm p值 > 0.01的模型,参见STAR方法)。值得注意的是,我们发现22个个体可以被拟合为伊朗农民相关祖先和AHG相关祖先之间的双向混合,而不含草原牧民相关祖先(称为ASI)。
印度外围梯度的考古背景及其与古代印度文明(如印度河谷文明)的关系尚不清楚,因为这些是来自中亚青铜时代文化的迁徙个体。此外,与现代印度人一样,他们也有一部分来自AHG相关群体的祖先。30 为了确定22个ASI个体和印度外围西部中伊朗农民相关祖先的最接近代表,我们使用了qpAdm并检查了从新石器时代到铁器时代的14个古代伊朗相关群体。当伊朗相关祖先来源于中亚文化的早期新石器时代和铜器时代个体时,我们对所有22个ASI个体都获得了良好的拟合——来自塔吉克斯坦的公元前4千年农民和牧民(Sarazm_EN,约公元前3,600-3,500年)或土库曼斯坦的铜器时代个体(Namazga_CA,约公元前3,300-3,200年)或Sarazm_EN和Parkhai_Anau_EN(约公元前3,500-2,900年,土库曼斯坦)的组合,这些之前被认为是印度外围梯度的来源30(数据S1,表S4.2)。对于印度梯度上的个体(n = 2,126),我们发现使用Sarazm_EN的模型为绝大多数(>95%)个体提供了最佳拟合(对于与AHG相关和Central_Steppe_MLBA的三路模型,p值 > 0.01)。相比之下,使用Namazga_CA或Parkhai_Anau_EN的模型对相当一部分(>15%)个体会失败或产生负系数(数据S1,表S4.3)。
转向印度梯度之外的个体(n = 494),我们尝试了三种模型,包括Sarazm_EN、AHG相关,以及(a)草原牧民相关(如印度梯度)、(b)南亚语系相关(使用尼科巴人)或(c)东亚相关(使用汉族)祖先。对于模型(b和c)失败的情况,我们还通过添加草原牧民相关祖先测试了四路模型。我们为494个个体中的91%获得了良好的拟合(数据S1,表S4.4)。在这些个体中,91个个体可以被模拟为不含草原牧民相关祖先,包括南亚语系相关群集中的几乎所有(∼96%)个体(使用模型b)。这表明,在采样人群中,印度ANI、ASI、南亚语系相关和东亚相关个体中伊朗农民祖先的最接近代表是Sarazm_EN。事实上,两个Sarazm_EN个体中的一个有一些AHG相关祖先的初步证据,正如先前所建议的37(数据S1,表S4.5和图S4.12)。
使用AHG相关、Sarazm_EN和Central_Steppe_MLBA作为参考群体,我们推断了印度梯度上个体的祖先比例。我们发现印度各地的遗传构成存在明显差异,AHG相关祖先在∼19%至69%之间变化,Sarazm_EN在∼27%至68%之间,而Central_Steppe_MLBA在∼0%至45%之间。在这三个祖先成分中,AHG相关变异与PCA中的ANI-ASI梯度显示出最强的相关性(数据S1,图S4.14)。AHG相关祖先比例与地理位置(例如,在印度南部最高,北部最低)、语言(即,在德拉维族语言使用者中高于印欧语系使用者)和社会群体(在部落群体中最高,与其他群体相比)显著相关,尽管每个群体内部存在较大变异(图1C)。这凸显了这些古代混合事件对印度遗传多样性产生了重要影响。
创始者事件增加印度的纯合性
先前的研究表明,许多印度群体由于内婚(社区内部婚姻)和近亲婚姻(近亲之间的婚姻)而有强烈创始者事件的历史。8,28,38 此类事件减少了遗传变异,降低了选择清除有害变异的效率,并增加了隐性疾病的风险。在基因组水平上,创始者事件增加了从少数共同祖先遗传的同源染色体区域(IBD)的共享。39 近亲婚姻后代更可能从父母双方继承IBD片段,导致同源隐性(HBD)片段。创始者事件导致许多短HBD片段,而近期近亲婚姻则导致数量较少但更长的HBD片段。
我们使用hap-IBD,40一种基于单倍型的IBD检测方法,在LASI-DAD和1000G数据集中识别了IBD和HBD片段。为了区分创始者事件和近期近亲婚姻的影响,我们按长度对HBD片段进行了分层——长片段(>8 cM)表明近亲婚姻,短片段(<8 cM)主要反映创始者事件。41 与1000G东亚人群(∼6 cM)、欧洲人群(∼6 cM)和非洲人群(∼4 cM)相比,印度人的基因组中平均有更大比例位于HBD片段中(∼29 cM)(图2A)。在印度内部,南部地区的个体具有明显更高的纯合性,无论是在基因组中HBD片段的总量方面(南部平均∼56 cM,而其他地区为∼19 cM,p值<10−16),还是在长HBD片段的比例方面(8.4%对4.3%,p值<10−6)。这反映了印度南部近亲婚姻更为普遍42(图2A;数据S1,图S5.1和S5.2)。大多数(>90%)纯合性来源于短HBD片段(而非长HBD片段),这表明历史创始者事件而非近期近亲婚姻是纯合性的主要来源(图2A和数据S1,图S5.2)。当我们使用20 cM作为阈值来定义长HBD片段时,获得了类似的结果(数据S1,图S5.1和S5.2B)。
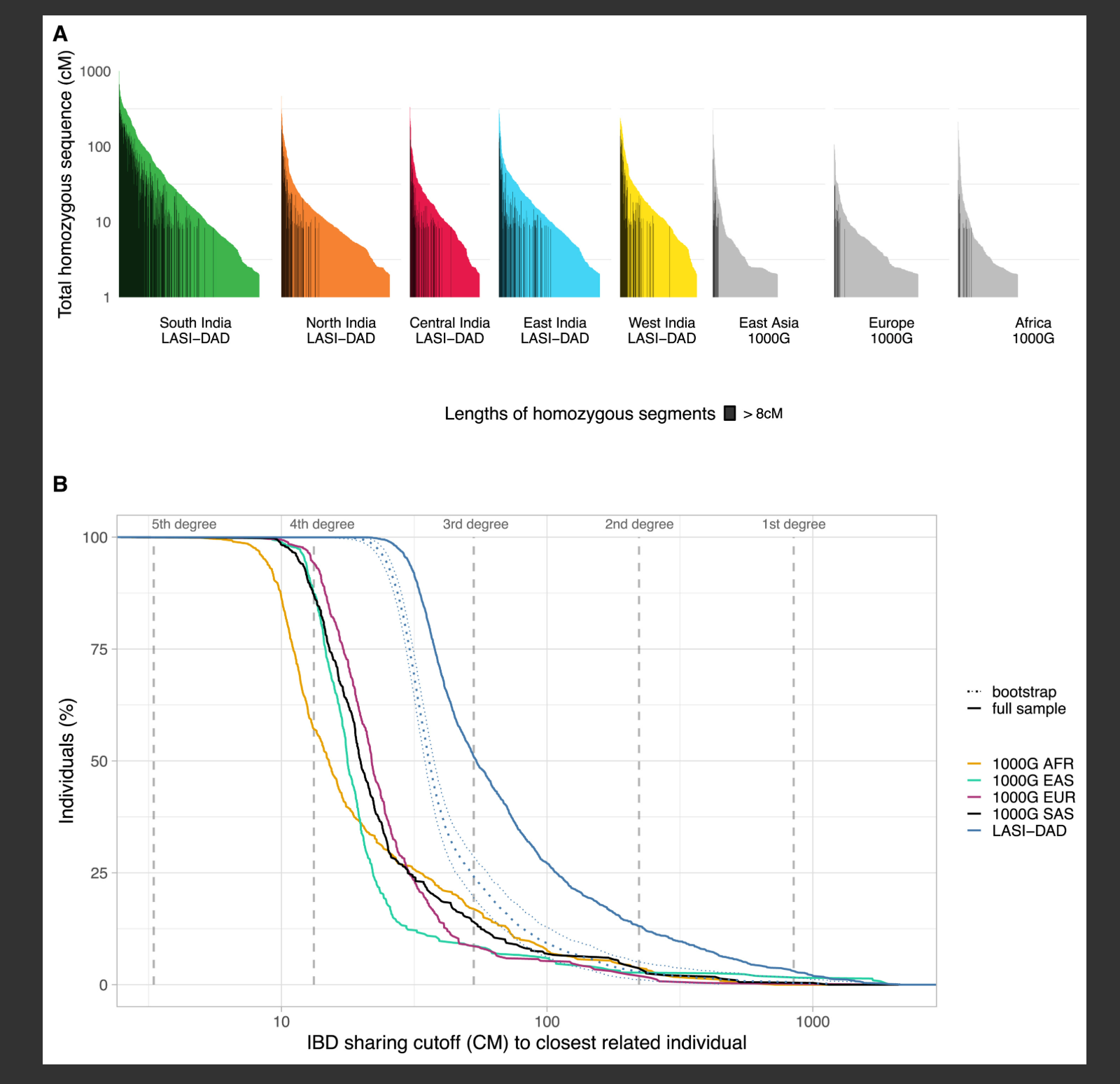
图2 创始者事件和近亲婚姻导致印度人较高的纯合性和亲缘关系
(A) 按地区划分的LASI-DAD样本全基因组纯合性,与来自东亚、欧洲和南亚的1000G个体相比。黑线显示>8 cM的纯合片段,彩色线包括较短片段。
(B) 对于LASI-DAD和1000G中的每个个体,我们识别了"最亲近的相关个体",即共享最高总量同源祖先(IBD)片段的个体,以厘摩(cM)为单位测量。y轴显示共享≥x cM的个体百分比(x轴)。对于LASI-DAD个体,我们通过重采样500个个体推断了均值和标准误差(虚线代表均值和95%置信区间)。垂直虚线表示k级表亲的总IBD共享的预期值。该图改编自Saada等人研究。
接下来,我们研究了个体间全基因组IBD共享情况。我们计算了LASI-DAD中至少有一个近亲的个体比例,并与1000G中的全球人群进行了比较(参见STAR Methods和Data S1,图S5.3)。我们推断LASI-DAD中约51.0%(各地区间为38.4%-59.2%)的个体至少有一个遗传关系相当于三度表亲或更近关系(约53 cM)的亲属,这明显高于1000G中AFR的17.2%、SAS的14.2%、EAS的8.8%和EUR的8.8%(图2B和Data S1,表S5.1)(注意先前研究发现在曼丁卡族冈比亚人[GWD]和尼日利亚埃桑人[ESN]中约5%-10%的个体是一度和二度亲属,这导致AFR中较高的亲缘关系44)。
LASI-DAD中较高的IBD共享,特别是与1000G SAS相比,可能源于:(1) LASI-DAD样本量更大,或(2) 两项研究中选择个体的抽样偏差。我们依次检验了这些假设。我们对LASI-DAD和1000G SAS中相同数量的个体(n = 500)进行了自举重采样,推断出三度表亲的比例降至24.2%(95%置信区间:19.4%-28.6%),但仍显著高于1000G SAS(图2B和Data S1,表S5.1)。在LASI-DAD中,个体采用分层抽样方案招募;首先在每个州内选择二级抽样单位(SSU)——村庄或城市人口普查区,然后在每个SSU内随机选择个体(见STAR Methods)。
为了控制我们样本选择方法的影响,我们比较了LASI-DAD中来自不同SSU的个体对(Data S1,第5节)。尽管控制了抽样位置和样本大小,我们仍发现LASI-DAD中的亲缘关系较高(约16.4%-35.0%),相比于1000G SAS(14.2%),尽管差异更为温和(Data S1,图S5.4)。这一比较凸显了1000G群体抽样在代表印度遗传变异方面的局限性(主要只有一些来自次大陆的外侨群体)。总体而言,我们发现LASI-DAD中的所有个体在我们的数据集中至少有一个可能的四度表亲或更近的亲属(IBD > 10 cM)。
印度古人类基因流
大多数非非洲人,包括印度人,其祖先中有约1%-2%来自古人类(尼安德特人和丹尼索瓦人)的基因流。5,7,45 然而,古人类祖先在印度的功能影响和区域变异仍不清楚。我们将一种称为hmmix的无参考隐马尔可夫模型33应用于印度的2,679个分相个体(为了最大化我们的样本量,我们保留了一级亲属[除了三人组的后代])。hmmix通过比较在外群中未发现的衍生等位基因密度(在这里,我们使用490个撒哈拉以南非洲人,他们具有可忽略不计的古人类祖先25),将基因组片段分类为两种状态——"现代人类"或"古人类"(参见STAR Methods)。为了将印度人的古人类祖先模式与全球其他人群进行比较,我们还将hmmix应用于1000G的2,309个个体和HGDP的825个个体的分相数据,并使用deCODE遗传学发表的hmmix对27,566名冰岛人的结果。26 除非另有说明,我们保留后验概率大于0.8的古人类祖先片段,在模拟中这相当于<4%的假阳性率。26
我们推断印度人的可检测基因组中平均有102.98 Mb或2.02%(95%百分位范围:1.79%-2.29%)来自古人类祖先。通过将推测的古人类片段与测序的尼安德特人和丹尼索瓦人基因组进行比较,46,47,48,49我们基于古人类片段上共享的衍生古人类变异(DAV)数量推断了古人类祖先的来源。我们发现每个个体有约1.43%(1.26%-1.65%)的尼安德特人祖先和约0.10%(0.03%-0.17%)的丹尼索瓦人祖先。印度的尼安德特人祖先比例与欧洲人(约1.2%)和美洲人(约1.3%)相似,但显著低于东亚人(约1.7%,Wilcoxon秩和检验p值<10-15)。丹尼索瓦人祖先比例最高的是大洋洲人(约2.0%),而美洲人、东亚人和南亚人的比例相似(约0.1%)(表S6)。
图3 印度和全球人群中的古人类基因流
现代人群中(A)尼安德特人和(B)丹尼索瓦人祖先的古人类祖先序列的Upset图和累积量(以Gb计)。为了与deCODE50进行比较,我们使用了严格的后验概率截断值(>0.9)并移除了重复区域中的任何SNP,以及(C)每个点代表使用hmmix中现代人类状态的发射参数估计的与撒哈拉以南非洲人的最小合并时间。x轴显示了按区域着色的每个人群,灰色区域标记了该人群与撒哈拉以南非洲人合并时间的95%置信区间。虚线表示托巴火山爆发时间(74,000年前51),反映了非洲南部扩散的最小估计时间。另见图S1。
接下来,我们计算了印度人与1000G和HGDP数据集中其他全球人群之间共享的古人类序列量(即在相同基因组区域重叠)。我们发现81.2%的尼安德特人祖先在至少两个全球区域之间共享(图3A),而约11.7%(1,679 Mb中的195.9 Mb)仅在印度观察到。总体而言,全球约90.8%的尼安德特人序列在印度人中被观察到(图S1)。即使在对样本量进行下采样以匹配1000G(n = 490)和HGDP(n = 28)中的最小样本量后,我们发现印度存在最大比例的尼安德特人祖先(n = 490时为84.5%[Data S1,图S6.9]和n = 29时为57.3%[Data S1,图S6.10])。此外,大洋洲人和印度人拥有大量与其他全球人群不共享的丹尼索瓦人祖先序列(Data S1,图S6.6)。约51%的丹尼索瓦人序列(591 Mb中的301.6 Mb)仅在印度人中观察到(Data S1,图S6.6),即使在下采样后这一点仍然显著(Data S1,图S6.8)。
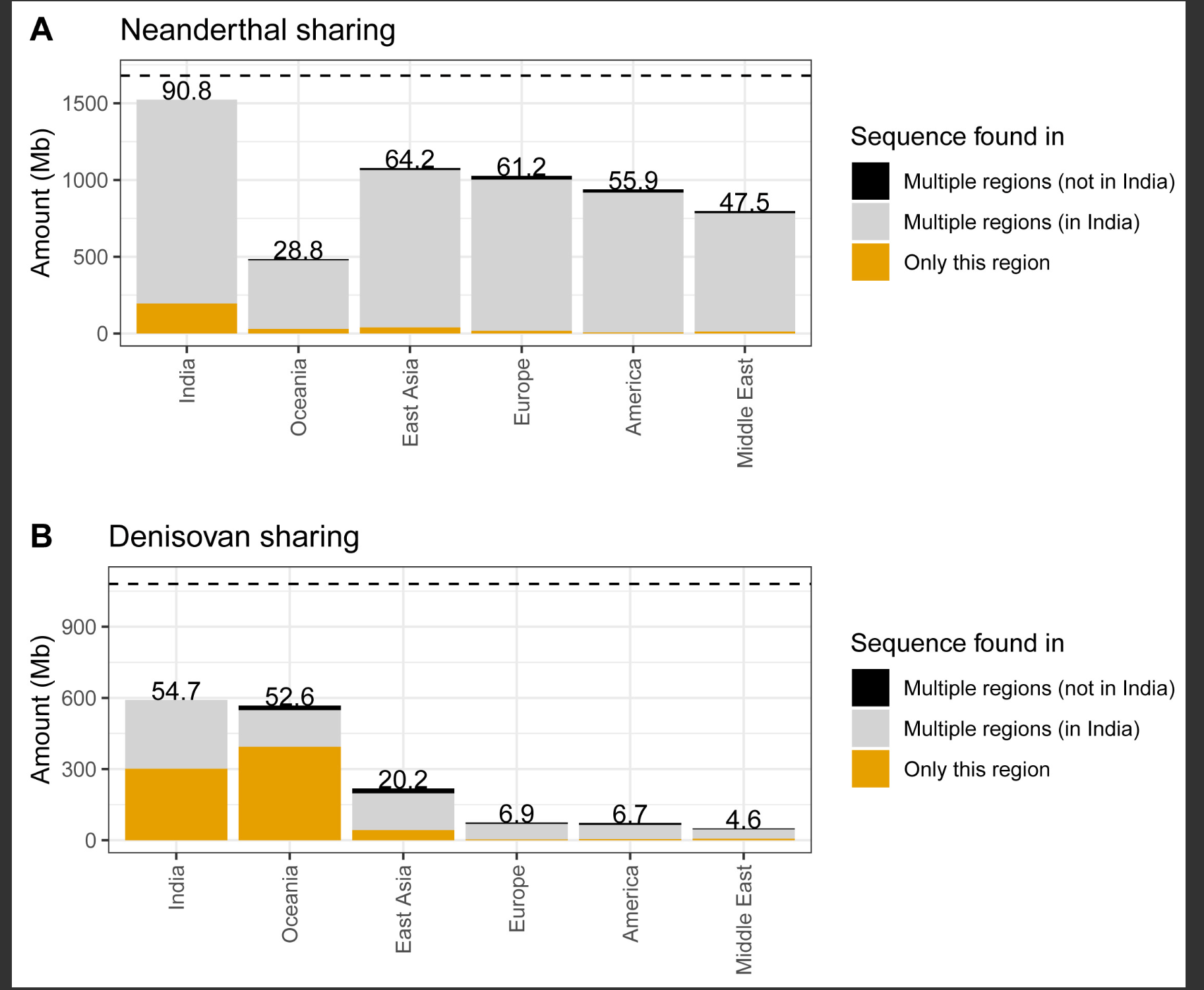
图S1 LASI-DAD、1000G和HGDP中全球个体发现的尼安德特人和丹尼索瓦人祖先序列共享情况,与图3相关
(A) 在后验概率截断值为0.8时发现的尼安德特人序列量(y轴),这些序列可能是任何区域特有的,或在多个区域(至少两个)中发现,其中一个包括印度(LASI-DAD数据集),或在多个区域(至少两个)中发现,但不包括印度。
(B) 与(A)相同,但针对丹尼索瓦人序列。水平线表示使用LASI-DAD、HGDP和1000G数据集组装的尼安德特人(1,679 Mb)和丹尼索瓦人(1,080 Mb)基因组的总长度。条形图上方的数字是占尼安德特人或丹尼索瓦人序列总量的百分比。
为了推断渗入的古人类群体与测序的古人类基因组之间的关系,我们估计了每个渗入片段中的DAV SNPs与三个高覆盖率尼安德特人46,47,48和一个丹尼索瓦人基因组49的匹配率。使用与Browning等人52类似的方法,我们发现平均而言,渗入的尼安德特人片段与三个测序的尼安德特人基因组之一共享83%的DAV(与Vindija尼安德特人共享最高),这重复了印度存在单次尼安德特人基因流的发现(Data S1,第6节)。印度大多数群体的丹尼索瓦人相关祖先来自一个与测序的阿尔泰丹尼索瓦人基因组关系较远的单一群体(共享46%-50%的DAV SNPs)。印度东北部和南部个体中的一小部分丹尼索瓦人相关祖先来自一个与测序的丹尼索瓦人基因组关系密切的丹尼索瓦人群体(片段平均共享84%的DAV SNPs)(Data S1,图S6.11)。印度东北部的个体携带来自东亚相关群体的近期祖先(图1B),这些群体先前已被证明携带两次丹尼索瓦人祖先脉冲。52 除了尼安德特人和丹尼索瓦人祖先外,我们推断印度人有0.42%(95%百分位范围:0.37%-0.48%)的古人类祖先来自未知来源(表S6)。这一比例在所有非非洲人中相似,可能与测序的古人类基因组和渗入的古人类个体之间的差异有关(Data S1,图S6.3)。因此,与先前的主张相反,没有明确证据表明其他未知古人类对印度人有额外贡献(至少不比其他全球人群多)。53
古老祖先在印度各地区有所不同,印度东北部和东部地区古老祖先比例最高,而北印度地区最低(Data S1,图S6.3;以及表S6.4和S6.6)。为了研究最近的基因流事件如何塑造印度古老祖先的分布,我们检查了尼安德特人和丹尼索瓦人祖先与印度三个主要祖先成分之间的关系。聚焦于印度谱系上的个体(n = 2,126),我们发现与AHG相关的祖先与丹尼索瓦人(r = 0.46,p值 < 10−15)和尼安德特人(r = 0.24,p值 < 10−15)祖先都呈正相关(Data S1,表S6.4)。通过使用更严格的标准来将古老祖先片段归属于尼安德特人和丹尼索瓦人起源,即使用只有一个古老群体具有与现代人类匹配的衍生等位基因的位点,这些结果是稳健的(Data S1,表S6.4)。这种模式,特别是丹尼索瓦人和AHG相关祖先之间的相关性,考虑到西欧亚相关群体具有最小的丹尼索瓦人祖先,这并不出人意料。54
印度次大陆走出非洲迁徙的时间
印度人口形成的一个核心问题是现代人类何时从非洲首次抵达次大陆。考古证据表明印度在约74,000年前发生的托巴火山爆发前后都有人类居住。51 然而,目前尚不清楚托巴火山爆发前的人群是否对当今印度人的祖先有所贡献。为了推断印度人与撒哈拉以南非洲群体的分离时间(即走出非洲迁徙的时间),我们使用了由hmmix推断的排放率(针对"现代人类"状态)。理论上,这一参数反映了个体自从与撒哈拉以南非洲人分离以来积累的突变数量。对于给定的突变率,我们因此可以推断与撒哈拉以南非洲人的最小合并时间(见STAR方法)。
此外,我们还检查了印度人推断的合并时间是否与东亚人、欧洲人和美洲人等其他非非洲群体相似(使用每个人群的hmmix排放率)。我们控制了跨人群和数据集的技术因素,包括相位错误、三等位基因位点的移除,并排除了任何具有超过1%撒哈拉以南非洲相关祖先的个体(因为这些因素都可能影响排放率)。假设人类突变率为每年每碱基对0.45 × 10−9,55 我们推断印度人与撒哈拉以南非洲人之间的最小合并时间是53,932年前(95%百分位范围:53,190–54,644)(Data S1,表S9.2;图3C;Data S1,图S9.3)。
我们在HGDP数据集中对欧洲人、东亚人和南亚人获得了质上相似的结果。此外,通过进行模拟,我们表明印度观察到的排放参数与来自约74,000年前较早迁徙的0%至3%祖先的变异一致(Data S1,图S9.5)。我们的结果表明,当今印度人的大部分祖先来源于约50,000年前发生的一次主要走出非洲的迁徙事件。
进化历史对疾病和功能变异的影响
人口历史,包括基因流动、创始者效应和自然选择,在塑造遗传变异(包括疾病易感性)方面发挥着关键作用。为了研究进化历史对印度遗传变异的影响,我们表征了变异的功能效应,包括那些改变蛋白质结构的变异,如推定功能丧失(pLoF)或错义变异(见STAR方法)。我们鉴定了385,985个错义变异和20,319个pLoF变异(Data S1,表S5.2)。每个个体在常染色体上携带约10,344个(范围:9,911-10,761)衍生错义变异和约67个(46-96)pLoF变异,与在其他全球人群中观察到的估计值相似。23
这些变异中的大多数(>90%)是罕见的(频率低于1%)或单例(约62%)。在人类基因组(RefSeq数据库56)的18,451个常染色体蛋白质编码基因中,我们在89.5%的基因中发现错义变异和pLoF(48%的基因中有pLoF),每个基因从1到1,265个变异不等(1-52个pLoF)。具有最多pLoF变异的前三个基因是黏蛋白基因:MUC3A、MUC16和MUC17,分别有52、42和41个pLoF,包括MUC17中的纯合pLoF。由于黏蛋白基因功能上存在部分冗余,可能对功能丧失变异有更大的耐受性。57
创始者事件历史预示着有害变异的高负担和隐性疾病风险的增加,如在芬兰人和阿什肯纳兹犹太人中所见。39,58 我们研究了印度个体中纯合有害突变负担(测量为纯合错义和pLoF的总和)的流行率变异。我们发现,与其他祖先相比,具有较高AHG相关祖先的个体携带更大的纯合有害突变负担(图4A)。值得注意的是,纯合有害突变负担与每个个体的HBD片段总和强烈相关。反过来,这意味着具有较高AHG相关祖先的个体中较高的突变负担是由较高的HBD驱动的,这是最近创始者事件和近亲婚配的结果(图4B)。在406,304个错义变异和pLoF中,我们发现约40%未在gnomAD或1000G中注册,这些变异的绝大多数极为罕见(<0.1%)(Data S1,表S5.2和图S5.5B)。我们发现约7%的非单例错义/pLoF变异存在于ClinVar数据库中,59 包括214个被分类为"致病性"或"可能致病性"的变异(Data S1,表S5.2)。许多ClinVar致病变异位于与罕见隐性疾病相关的基因中,如与血液疾病相关的HBB,60 与先天性听力丧失相关的GJB2,61 与囊性纤维化相关的CFTR,62 以及在苯丙酮尿症中发挥作用的PAH。63 值得注意的是,我们在丁酰胆碱酯酶(BCHE)基因中发现了一个致病变异(L307P),该变异存在于LASI-DAD中的15个个体(0.28%)中,但在南亚以外不见64(Data S1,表S5.2)。BCHE缺陷的患者在使用麻醉过程中常用的某些肌肉松弛剂时,肌肉麻痹风险较高。这种变异在生活在安得拉邦和特伦甘纳邦的Vysya社区个体中富集,28,64,65 这与我们的观察一致,即LASI-DAD中15个携带者中有8个来自特伦甘纳邦。这些发现强调了特定人群遗传筛查计划在减轻印度疾病负担方面的价值。
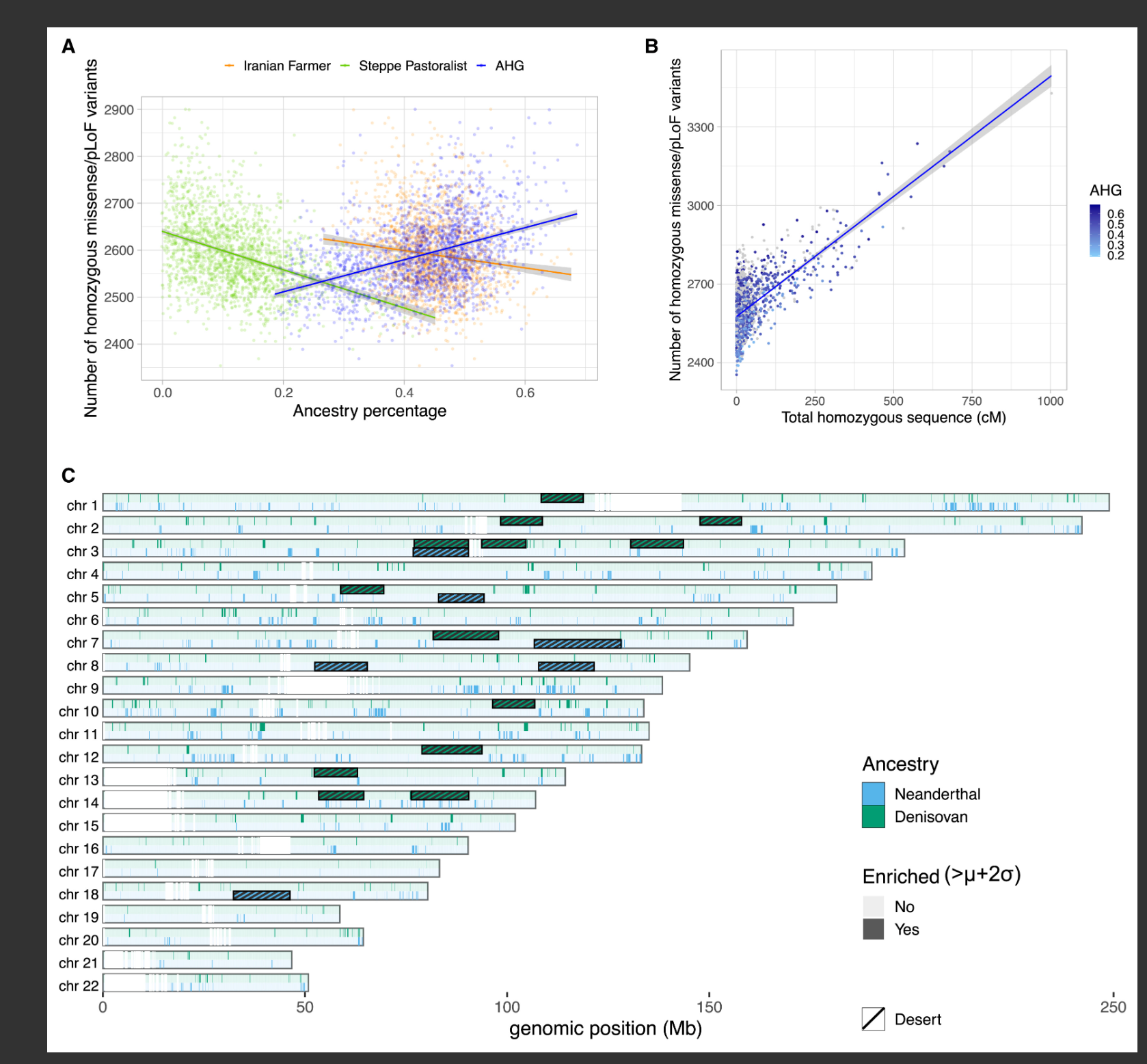
图4 人口统计学历史对疾病风险的影响
(A) 印度分化线上个体的纯合衍生错义/pLoF数量与祖先系数之间的关系。为更好地显示趋势,y轴被截断。
(B) 纯合衍生错义/pLoF数量与每个个体HBD片段总和之间的关系。个体根据其AHG相关祖先系数着色,不在分化线上的个体为灰色。我们使用广义线性模型(GLM)拟合回归,得到以下拟合:y = 2,576 + 0.916 × x。
(C) 古人类祖先区域在基因组中的分布。我们计算了LASI-DAD个体基因组上的平均古人类频率,并将具有高于平均值(μ)加两个标准差(σ)的古人类频率的片段视为富集区域(尼安德特人为蓝色,丹尼索瓦人为绿色)。古人类祖先沙漠(10 Mb区域内古人类祖先<0.1%)以相同颜色的条纹矩形显示。
为了描述古人类祖先在印度的功能影响,我们研究了古人类祖先在全基因组的分布,并确定了"高频古人类祖先"区域(定义为个体间古人类祖先频率高于全基因组平均值两个标准差的区域)(图4C)。我们分别鉴定了1,590个和818个具有高频尼安德特人和丹尼索瓦人祖先的候选区域。对于尼安德特人,我们重复验证了先前确定的基因,如FBP2和FYCO1,并确定了PCAT7和CXCR6作为额外的候选基因。对于丹尼索瓦人,我们重复验证了WDFY2、CHD1L和HELZ2中的信号,并确定了几个额外的候选基因,包括LINC00708和CDKN2B(Data S1,第7节;表S3)。通过基因本体论(GO)富集分析,我们发现14个富集尼安德特人祖先和22个富集丹尼索瓦人祖先的通路,主要与免疫功能相关(表S4)。
接下来,我们寻找具有大量现代人类和古人类群体共享的衍生等位基因的区域,这种特征先前在EPAS1基因和西藏人的丹尼索瓦人祖先中被观察到。有趣的是,我们发现基因组的一个区域具有不成比例地高的仅与印度人共享的丹尼索瓦人衍生变异;然而,尼安德特人衍生变异没有表现出类似的富集(Data S1,第7节)。这个包含BTNL2基因的区域,是主要组织相容性复合体(MHC)的一部分,在13.2千碱基(kb)区域内包含78个丹尼索瓦人特异性衍生变异。它在印度人中也具有特别高(约10%)的丹尼索瓦人祖先(>99.9百分位)。这个区域的丹尼索瓦人单倍型按长度聚类:一个55-65 kb的短单倍型和一个约150 kb的长单倍型,分别包含116.1和126.7个丹尼索瓦人特异性衍生变异。单倍型长度和印度人与丹尼索瓦人之间共享的衍生等位基因数量表明,这个区域可能是来自丹尼索瓦人或丹尼索瓦人相关人群的基因流的产物,而非祖先谱系分选(长单倍型的p值<10-6;短单倍型的p值=0.027)。在全球人群中,这些丹尼索瓦人单倍型在东亚人中也呈现高频率(约11.8%,>99.8百分位),但在欧洲人中很罕见(约0.4%),并且值得注意的是,在大洋洲人群中缺失(Data S1,表S7.2)。MHC包含许多与免疫功能和传染病相关的基因,这些基因可能受到平衡选择的影响。事实上,先前的研究已经确定BTNL2在东亚人中是平衡选择的候选基因。尽管模拟表明,仅由强烈的创始者事件产生的遗传漂变可以迅速改变古人类祖先的频率(即使在没有选择的情况下),因此高频古人类祖先区域的解释应谨慎(Data S1,第8节)。
为了确定在印度人中富集的古人类祖先区域(与东亚人和欧洲人相比),我们计算了群体分支统计(PBS)。PBS测量自人口(印度)与两个参考群体分离以来频率的增加。对于每个人群,我们测量了尼安德特人和丹尼索瓦人祖先在基因组窗口中的频率(见"STAR方法")。我们确定了约10.7 Mb(或235个基因)富集尼安德特人祖先和约5.5 Mb(或84个基因)富集丹尼索瓦人祖先的区域(Data S1,图S7.5C;表S3)。丹尼索瓦人富集区域包含与先天免疫反应相关的基因,包括几个TRIM基因—TRIM26、TRIM31、TRIM15、TRIM10和TRIM40—这些基因与病毒进入(或退出)宿主细胞的细胞过程有关。尼安德特人最显著富集区域之一包括位于染色体3上的一个基因簇,先前已与SARS-CoV-2感染导致的呼吸衰竭风险增加有关(PBS尼安德特人>0.118,在全基因组PBS分数的99.99百分位)。这个区域有两个主要的尼安德特人起源单倍型:一个49.4 kb的核心单倍型和一个333.8 kb的长单倍型(Data S1,图S7.6A和S7.7)。在印度各地,核心单倍型的频率从东北部的20.5%到东印度的34.8%不等。核心和长单倍型在印度东部的频率明显高于其他地区(核心,34.8%,Z=2.68;长,23.2%,Z=2.34,Data S1,表S7.3;Data S1,图S7.6B)。我们还发现39个超过1 Mb的非常长的单倍型。
我们还检查了现代人类基因组中缺乏古人类祖先的区域,称为"古人类沙漠"。我们识别了六个尼安德特人沙漠,总跨度为87.1 Mb,其中包括五个先前已报道的(图4C;Data S1,图S7.8和表S7.4)。我们还在印度人中识别了13个丹尼索瓦人沙漠,其中两个与先前报道的尼安德特人沙漠重叠(图3C;Data S1,图S7.9和表S7.5)。考虑到印度人基因组中丹尼索瓦人祖先比例较低,额外的取样可能会在这些沙漠中发现丹尼索瓦人祖先。先前的研究表明,在基因附近,特别是在基因组的功能重要区域,古人类祖先的减少更为明显,这表明杂交变异是有害的,并被净化选择所清除。因此,我们检查了18,451个蛋白质编码基因中古人类祖先的频率,发现尼安德特人祖先在基因之间的变化范围为0%至36.7%,丹尼索瓦人祖先为0%至21.9%。许多基因(约35%)不含有古人类祖先片段(Data S1,图S7.10),这在我们方法的分辨率限制内。这些基因中包括载脂蛋白E(APOE)基因,该基因携带APOE ε4变体,这是晚发性阿尔茨海默病(AD)的主要风险因素。然而,根据我们目前的样本量,这些结果并不显著(单侧经验p值 = 0.16),应在更大队列中重复验证(Data S1,图S7.10)。
讨论
印度是一个拥有非凡遗传多样性的地区。我们已经生成了对印度遗传变异最全面的调查,包括来自大多数地理区域、所有主要语言家族的使用者以及部落和历史上代表性不足的社区。我们表明,印度人的大部分遗传变异源于约50,000年前发生的一次主要走出非洲的迁徙,随后有来自尼安德特人和丹尼索瓦人的基因流。如今,大多数印度人拥有来自三个主要来源的祖先,这些来源与南亚狩猎采集者(AHG)、古代伊朗农民和欧亚草原牧民群体有关。通过检查新石器时代至铁器时代的14个古代伊朗群体的数据,我们发现了一个共同的伊朗农民祖先来源,这与公元前4千年的塔吉克斯坦农民和牧民(Sarazm_EN,约公元前3,600-3,500年)有关,这些祖先进入了ASI、ANI、与南亚语系相关和与东亚相关的印度群体的祖先。考古研究也记录了萨拉兹姆(Sarazm)与南亚之间的贸易联系,包括与梅赫尔加尔(Mehrgarh)农业遗址和早期印度河流域文明的联系。74事实上,两个Sarazm_EN个体中的一个被发现佩戴贝壳手镯,这些手镯与在巴基斯坦和印度的遗址如沙希-图姆普(Shahi-Tump)、马克兰(Makran)和古吉拉特的苏尔科塔达(Surkotada)发现的手镯完全相同。75在这些混合之后,印度经历了向内婚制的重大人口转变,导致个体间广泛的纯合性和相同祖先共享。
印度人的高度亲缘关系值得注意:在约2,700人的样本量中,我们发现每个个体在我们的研究中至少有一个推定的四等亲或更近的亲属(共享的IBD量>10 cM)。76此外,印度人的纯合性水平升高(各地区平均范围从每个个体约12到56 cM不等),是东亚人和欧洲人的二到九倍。这些发现强调了印度人之间广泛的家族联系,反映了历史、文化或社会模式,如内婚制。纯合性增加了有害变异的流行和隐性疾病的风险。我们生成了错义和推定功能丧失变异的目录,发现超过160,000个变异(未在之前的基因组调查中登记)。正如我们最近发表的文章所报道的,这些变异中许多在ClinVar中有注释,并与先天性和血液疾病、代谢疾病和药物反应,以及认知衰退和痴呆等复杂疾病相关。77值得注意的是,这些变异在印度以外没有被观察到,在整个印度的频率较低,但在某些社区中相当常见,例如导致BCHE缺乏症的致病性错义变异(L307P)的分布所示。正如在阿什肯纳兹犹太人和芬兰人的研究中先前所示,这些变异的识别和映射对于提高我们对疾病病因的理解和减轻疾病负担具有巨大潜力。39,58
在深层时间尺度上,印度人携带1%-2%来自古老人类、尼安德特人和丹尼索瓦人的祖先血统。印度人在现代人类中展示了最大的古人类祖先变异。值得注意的是,如今现代人中存在的尼安德特人祖先血统大部分在印度被发现,而全球其他人群仅保留了这种变异的一个子集。我们发现来自尼安德特人和丹尼索瓦人的基因渗入变异对适应和疾病有所贡献。几种古人类变异在参与免疫功能的基因和通路中富集,包括丹尼索瓦人遗传的单倍型在MHC复合体中富集,与先天免疫反应相关的TRIM家族基因,以及第3号染色体上尼安德特人遗传的基因簇,其中包含SARS-CoV-2感染后严重症状的主要风险因素。利用这些知识可以帮助开发针对印度人群量身定制的新型治疗方法,特别是针对具有免疫相关成分的疾病,如自身免疫性疾病和传染病。我们识别了六个尼安德特人和13个丹尼索瓦人祖先沙漠,包括四个既缺乏尼安德特人又缺乏丹尼索瓦人祖先的区域。有趣的是,其中一个区域包括FOXP2基因,该基因与人类语言发展相关。对这些沙漠的功能研究可能会发现先前未表征的遗传变异,这些变异有助于现代人特有的特征和疾病。
总之,这些发现提供了印度遗传景观的全面视图,强调了次大陆上深层进化历史、人口变迁以及古代和近期基因流事件对遗传变异的影响。印度人独特的遗传结构凸显了在未来医学和功能基因组学研究中纳入祖先和纯合性的重要性。
研究局限性
在这项研究中,我们比较了来自印度约2,700个个体的全基因组序列与其他全球人群,包括古代和当代个体。利用这些数据,我们描述了不同个体的祖先构成,并研究了人口统计学历史对印度的遗传变异和疾病的影响。我们的结果依赖于来自不同人群和时间尺度的参考个体的可用性。例如,我们确定印度伊朗农民相关祖先的最接近代理(在采样个体中)是来自中亚文化的公元前4千年的农民和牧民。然而,我们来自这一地区和时间尺度的遗传数据非常稀少,随着更多来自印度和中亚的古DNA数据变得可用,重新审视这些分析以确认印度伊朗农民祖先的来源将变得重要。此外,使用局部祖先推断方法识别现代印度人基因组中的狩猎采集者、伊朗农民和草原牧民相关祖先区域,将揭示次大陆上环境适应和疾病易感性的来源和动态。
 陕公网安备61011302002223号
陕公网安备61011302002223号